


Radical cystectomy is the standard treatment for infiltrating bladder cancer. This surgery is associated with a high morbidity due to structure resection and to metabolic complications derived from urinary diversion, such as hyperammonemic encephalopathy, an uncommon complication that may occur several years after the surgical procedure.1
The case of a 78-year-old female patient with an unremarkable personal and family history diagnosed 5 years earlier with grade III, stage B bladder carcinoma treated with cystectomy, hysterectomy, double adnexectomy, and ureterosigmoidostomy is reported.
Four years after surgery, she was admitted to another hospital for episodes of disconnection from the environment, sucking movements, myoclonic twitching in the head and right limbs, episodes of amnesia, and postictal confusion. Magnetic resonance imaging showed normal results, and an electroencephalogram showed marked bilateral frontotemporal activity. Idiopathic epilepsy was diagnosed, and antiepileptic treatment was started (levetiracetam 500mg/12h).
Three months later she was admitted again to the same hospital with fever, and hyperchloremic metabolic acidosis was found. Because of prior urinary diversion, ammonia levels in blood were tested and were found to be 200μg/dL (normal range, 17–80μg/dL). The patient had no symptoms of chronic hyperammonemia during the intercritical period.
Tests for autoimmunity, hormones (TSH 2.36μU/mL [0.27–4.2] and free thyroxine 1.05ng/dL [0.93–1.7]), and tumor markers, viral serologic testing, and abdominal ultrasonography were performed to rule out a hepatic origin of hyperammonemia with normal results.
Based on a diagnosis of encephalopathy with a non-convulsive status of complex partial seizures of toxic-metabolic origin and hyperchloremic metabolic acidosis secondary to ureterointestinal diversion, the patient was managed with a low-protein diet, hydration with 2L/day, oral bicarbonate 500mg/8h, lactulose 10g/8h, and levetiracetam 500mg/12h, and was referred to the nutrition unit of our hospital for an adjustment of nutritional therapy.
The patient reported a weight loss of 8% since surgery. She weighed 55kg and had a body mass index of 27kg/m2, a tricipital skinfold of 18mm, arm circumference of 25cm, arm muscle circumference of 19.35cm, and grade B overall subjective assessment. Blood pressure values were 120/80mmHg, the physical examination was normal, and there was no ankle edema.
After work-up, a total restriction of proteins of animal origin and a supplementation of proteins of vegetable origin were recommended.
An evaluation of dietary intake by means of a dietetic diary on three non-consecutive days was requested. At the control visit, a daily intake of 40g of protein and 1300kcal was seen.
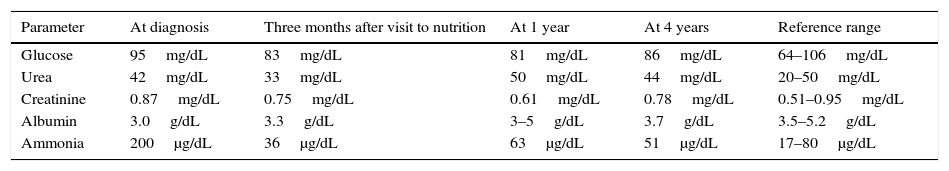
Table 1 shows blood test results at diagnosis, after the first visit to the nutrition clinic, and after 1 and 4 years.
Blood chemistry at diagnosis, after the first visit to the nutrition clinic, and 1 and 4 years after the start of nutritional therapy.
| Parameter | At diagnosis | Three months after visit to nutrition | At 1 year | At 4 years | Reference range |
|---|---|---|---|---|---|
| Glucose | 95mg/dL | 83mg/dL | 81mg/dL | 86mg/dL | 64–106mg/dL |
| Urea | 42mg/dL | 33mg/dL | 50mg/dL | 44mg/dL | 20–50mg/dL |
| Creatinine | 0.87mg/dL | 0.75mg/dL | 0.61mg/dL | 0.78mg/dL | 0.51–0.95mg/dL |
| Albumin | 3.0g/dL | 3.3g/dL | 3–5g/dL | 3.7g/dL | 3.5–5.2g/dL |
| Ammonia | 200μg/dL | 36μg/dL | 63μg/dL | 51μg/dL | 17–80μg/dL |
The elimination of proteins of animal origin was again emphasized, except for the occasional egg to supplement proteins of vegetable origin, and an individualized nutritional regimen was devised. Treatment with calcifediol 266μg/month was started when vitamin D deficiency was detected.
After 3 months of individualized diet, the patient had not been readmitted, had maintained her weight, and had normal ammonia levels (36μg/dL), kidney and liver function, and vitamin D levels.
Normal blood ammonia levels were found in all subsequent measurements, with no clinical or biochemical evidence of protein malnutrition.
In this patient, encephalopathy occurred because the bowel segments used for ureteral diversion retain their absorption and secretion capacity,2 which results in increased ammonia absorption with saturation of the metabolic capacity of the liver and hyperammonemia. Sodium and bicarbonate secretion, as well as the reabsorption of hydrogen ions and chlorine by the intestinal mucosa, causes hyperchloremic metabolic acidosis.3
Under normal conditions, ammonia mainly comes from the bowel, where it is generated by the metabolism of nitrogenated products in the diet, the action of intestinal flora, and glutamine metabolism by intestinal glutaminase. Ammonia is absorbed from the small bowel and reaches portal circulation, finally arriving at the liver, where 90% is metabolized by the urea cycle.4,5
Protein-rich diets and intestinal bacterial growth with a predominant proteolytic flora increase ammonia synthesis and availability for absorption.
In ureterointestinal diversion, ammonia absorption is promoted by the alkalinization of the bowel lumen, so that urinary ammonia is converted to its ionized form (NH4+), which is liposoluble and diffuses more readily through biological membranes,5 and increased ammonia production resulting from the overgrowth of proteolytic enzymes.6 In addition, as ammonia is absorbed by distal bowel segments draining to the inferior hemorrhoidal plexus, a direct passage into the systemic circulation with no liver metabolism occurs,7 promoting hyperammonemic encephalopathy.
The main treatment for hyperammonemia for non-hepatic causes is ammonia reduction in the bowel lumen. Treatments proposed include decreased exogenous protein provision and the use of non-absorbable disaccharides to decrease ammonia formation by flora and to promote intestinal excretion.
As regards protein, no agreement exists regarding the restrictions required by this disease. In the case reported, a protein provision of 0.8g/kg/day induced no clinical evidence of energy and protein malnutrition. Dietary recommendations were made to achieve maximum protein quality through the combined use of vegetable proteins, because cereals are deficient in lysine, corn is poor in tryptophan, and legumes are poor in methionine and cysteine. In addition, the fiber associated with these foods favors the growth of saccharolytic over proteolytic bacterial flora. The patient was also advised to eat eggs occasionally, as they contain protein of the highest quality. With these changes, the patient has maintained an adequate nutritional state with no reappearance of her neurological symptoms.
As regards drug treatment, non-absorbable disaccharides (such as lactulose) are metabolized by saccharolytic intestinal flora in the colon, generating short-chain fatty acids that decrease intestinal pH and, thus, ammonia absorption. Treatment with antiepileptic drugs for epileptic states secondary to toxic-metabolic causes in patients with no history of epilepsy or cerebral structural organic lesion may be gradually decreased if the patient is free from seizures after the complete correction of the underlying cause. If the recurrence of the metabolic impairment is expected, the prescribed drug may be maintained at the minimum effective dose as prevention.8
While uncommon, hyperammonemia for non-hepatic cause may be due to changes in the urea cycle, such as ornithine transcarbamylase (OTC) deficiency. The fact that a deficiency of this enzyme coded in chromosome X first becomes evident in adulthood is usually due to the concurrence in heterozygous subjects of triggering factors (infection, trauma, valproate administration, surgery, stress, or excess protein intake).9 Clinical suspicion of these deficiencies if neurological symptoms (mental retardation, ataxia, irritability, aggressiveness, confusion, hallucinations) and hyperammonemia occur in the same family is important.
If the clinical signs and symptoms persist, the conversion of urinary diversion to an ileal conduit may be considered, as this has been shown to normalize ammonia levels and to eliminate symptoms in a refractory patient.1
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Moriana M, Martinez-Ibañez J, Civera M, Martínez-Valls JF, Ascaso JF. Encefalopatía hiperamoniémica tras cistectomía radical y derivación urinaria. Tratamiento nutricional. Endocrinol Nutr. 2016;63:306–308.
