



Glucocorticoid resistance (GCCR, OMIM #615962) is a rare condition characterized by generalized, partial, target tissue resistance to glucocorticoids. Cortisol action mediated by the glucocorticoid receptor (GR) is decreased, with a compensatory elevation of circulating ACTH. This increases the production of glucocorticoids, mineralocorticoids and adrenal androgens.1 The diurnal rhythm of cortisol is maintained but in an elevated level and no adequate cortisol suppression is observed after 1mg dexamethasone (DXM) test.1
The molecular basis of GCCR has been related to mutations in the NR3C1 gene, located on 5q31–q32, which encodes for the GR (Fig. 1A).
 Schematic representation of the structure and functions of the human glucocorticoid receptor (GR). Exon 1 is not translated. AF-1, activation function-1 domain; DBD, DNA-binding domain; LBD, ligand-binding domain. (B) Schematic diagram of the zinc finger structure of the DBD of the human GR. Several residues at these zinc fingers are required for dimerization (aminoacids 458–462; highlighted in bold), transactivation (aminoacids 469, 470, 472; boxed) or to repress other transcription factors, such as AP-1 (aminoacids 425, 436, 478; circled). Aminoacid 477, affected in Case 1, is represented double-underlined.")
(A) Schematic representation of the structure and functions of the human glucocorticoid receptor (GR). Exon 1 is not translated. AF-1, activation function-1 domain; DBD, DNA-binding domain; LBD, ligand-binding domain. (B) Schematic diagram of the zinc finger structure of the DBD of the human GR. Several residues at these zinc fingers are required for dimerization (aminoacids 458–462; highlighted in bold), transactivation (aminoacids 469, 470, 472; boxed) or to repress other transcription factors, such as AP-1 (aminoacids 425, 436, 478; circled). Aminoacid 477, affected in Case 1, is represented double-underlined.
In this letter, we report two novel heterozygous mutations in the NR3C1 gene in two unrelated patients in whom glucocorticoid resistance was suspected.
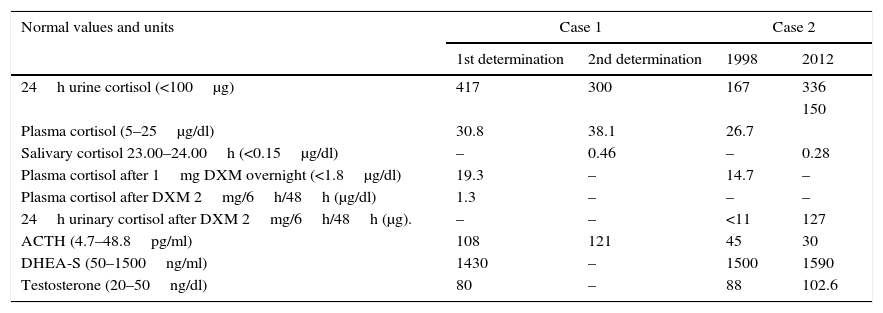
Biochemical values obtained from the patients are shown in Table 1. In both cases, these pointed to an ACTH-dependent Cushing's syndrome. No pituitary lesion was observed in either patient's magnetic resonance imaging (MRI). Bone densitometry was also normal in both cases.
Biochemical results of the patients. Biochemical values obtained in the different tests for the two patients. Normal values at the local laboratory are indicated in brackets in the first column. DXM, dexamethasone. DHEA-S, dehydroepiandrosterone sulfate.
| Normal values and units | Case 1 | Case 2 | ||
|---|---|---|---|---|
| 1st determination | 2nd determination | 1998 | 2012 | |
| 24h urine cortisol (<100μg) | 417 | 300 | 167 | 336 |
| 150 | ||||
| Plasma cortisol (5–25μg/dl) | 30.8 | 38.1 | 26.7 | |
| Salivary cortisol 23.00–24.00h (<0.15μg/dl) | – | 0.46 | – | 0.28 |
| Plasma cortisol after 1mg DXM overnight (<1.8μg/dl) | 19.3 | – | 14.7 | – |
| Plasma cortisol after DXM 2mg/6h/48h (μg/dl) | 1.3 | – | – | – |
| 24h urinary cortisol after DXM 2mg/6h/48h (μg). | – | – | <11 | 127 |
| ACTH (4.7–48.8pg/ml) | 108 | 121 | 45 | 30 |
| DHEA-S (50–1500ng/ml) | 1430 | – | 1500 | 1590 |
| Testosterone (20–50ng/dl) | 80 | – | 88 | 102.6 |
Patient 1 was a 12-years old girl referred to the endocrinologist after her pediatrician observed a clinically irrelevant white stretch mark on the right thigh and, afterwards, a high basal cortisol level. Menarche occurred at the age of 11 and she referred regular periods. Physical evaluation was normal (weight in percentile 75, height in percentile 90 and Tanner stage IV for pubertal development). Her blood pressure was normal. She presented mild hirsutism. Physiological causes for elevated cortisol levels were excluded. As she remained asymptomatic, no therapeutic attitude was taken. Subsequent evaluations showed no physical or biochemical changes. Hormone studies were performed in the patient's parents. Her mother had slightly elevated cortisolemia (27μg/dl), with normal DHEA-S, testosterone and 17-Hydroxyprogesterone values. Plasma cortisol level after 1mg oral DXM administration overnight was 3.7μg/dl. Father's results were normal.
Patient 2 was a 41-years-old woman referred to the endocrinologist for hirsutism in 1998. She presented regular menses with no other symptoms or signs of Cushing's syndrome. She was depressed after her mother's terminal disease. She chose electrolysis for permanent hair removal after unsuccessful medical treatment. In 2012, she claimed an increment of the hirsutism, chronic fatigue and anxiety. Physical examination was normal except for the hirsutism (weight 50.6kg, height 154cm, BMI 21.3kg/m2, BP 125/80mmHg). Diabetes was ruled out (fasting glucose, 85mg/dl; HbA1c 5%). Hormonal tests persisted altered.
Genetic analyses were performed after written informed consent and local ethical committee endorsement were obtained. Briefly, PCR amplification and direct sequencing of the nine NR3C1 gene exons were performed and sequences were compared with the reference sequence (Ensembl identifier: ENSG00000113580) using specific software. Primer sequences and conditions are available on request.
We identified two novel heterozygous mutations in the NR3C1 gene. Patient 1 and her mother presented the p.Arg477Cys (c.1429C>T) mutation, located in exon 4, in the second zinc finger of the DNA binding domain of the protein. Its ‘in silico’ functional effect was assessed using pathogenicity prediction software, being characterized as pathogenic. Despite this mutation is novel, another naturally occurring point mutation has been previously reported at the same amino acid position.2 The clinically severe p.Arg477His was related with the elimination of the transactivating activity and primary cortisol resistance, still presenting intact ligand binding capacity. DNA binding capacity was decreased. It also presented a negative effect on the transcriptional activity. It is probable that the p.Arg477Cys mutation, located in the same aminoacid, behaves the same way as p.Arg477His, affecting the transactivating activity and/or DNA binding capacity.
Patient 2 carried the p.His588Leufs*5 (c.1762_1763insTTAC) mutation, in exon 6, in the ligand binding domain (LBD) of GR. This frameshift mutation determines a substitution of four amino acid residues at codons 588–591 and a premature stop codon at position 592, thus leading to a truncated protein. The alteration is located at the second charge clamp of the steroid binding domain, and point mutations in this region have been described to reduce activation mediated by the LBD.3 In this case, lack of the C-terminal domain of the protein is expected to alter some functions of the LBD. Another similar truncated protein already described was predicted to have a dominant negative activity by impairing wild-type GR nuclear translocation. Similar features may be expected from the p.His588Leufs*5 mutation.
In summary, in this article we report two novel pathogenic mutations in the glucocorticoid receptor gene in two unrelated patients with GCCR. The studied patients presented an ACTH-dependent hypercortisolism with no symptoms, except hirsutism and, more recently, anxiety and chronic fatigue in Patient 2. Other causes of ACTH-dependent hypercortisolism were ruled out and the persistence in time of elevated cortisol and ACTH levels without any other clinical symptom drove us to the diagnosis of GCCR.
Generalized glucocorticoid resistance can have a variable clinical presentation, ranging from absence of symptoms to the presence of clinical features due to an excess of mineralocorticoids and/or adrenal androgens.1,4 Severity of the disease cannot be inferred from the type of mutation in the NR3C1 gene, as there is not a clear phenotype-genotype relationship, with clinical symptoms varying even among patients and relatives with the same disease-causing mutation.5 Therefore, GCCR may be suspected when hypercortisolism is present and there is no other associated clinical feature. Molecular analysis of the NR3C1 gene is recommended in order to confirm the diagnosis and provide a better genetic counseling to the affected families.
FundingTV is supported by the FPI Program of the University of Basque Country (UPV-EHU) (IF/UPV/12/135).
Conflict of interestThe authors have nothing to disclose.
