


Carney's complex is a syndrome first described in 1985 and is characterized by the presence of myxomas, skin hyperpigmentation, and endocrine overactivity. It is a rare autosomal dominant disease with variable expressivity of which approximately 600 cases have been reported worldwide. Although it has been related to two different genetic loci, inactivating mutations in the PRKAR1A are found in 70% of patients. These mutations occur de novo in 30% of patients.1
The case of a patient with acromegaly in the setting of CC is reported below.
This was a 55-year-old woman who was admitted to the cardiology department for left atrial myxoma and another probable myxoma on the right side. The patient experienced a seizure during admission, which led to magnetic resonance imaging (MRI) being requested. This revealed a 1.2cm pituitary tumor that displaced the pituitary stalk to the right but did not affect the suprasellar region and was not in contact with the chiasmatic region.
The personal history of the patient included a left atrial myxoma, complicated with several embolic strokes, detected at 31 years of age. The myxoma was resected but relapsed three years later, requiring repeat surgery. She had focal epilepsy as a sequela. At 39 years of age carcinoma in her left breast was detected. At the age of 45, she was evaluated at the dermatology department because of the occurrence of skin myxomas, nevi, and lentiginosis since adolescence. Several fibroadenomas were resected from her right breast at 54 years of age. A genetic study was then requested because of the clinical suspicion of CC. The result of this study was not available during the initial endocrinological assessment.
A targeted clinical history found marked hand and feet growth over a long period, with an increase in shoe size from 37 (at 25 years) to the current 41, and joint pain, mainly in the carpal joints. The patient had not noticed any voice changes, and had no high blood pressure or changes in carbohydrate metabolism. She reported hair increase in the scalp only. The menstrual rhythm of the patient had been regular until one year before consultation, and she reported no galactorrhea. A physical examination found acromegaloid facial stigmata, with macroglossia and marked soft tissue thickening. She also had lentiginosis in the neck and back, as well as neck skin myxomas.
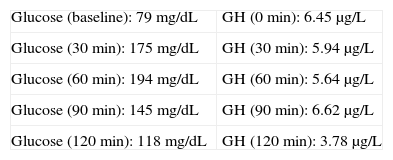
Baseline laboratory levels included: thyrotropin (TSH) 2.99μIU/mL (0.4–4), thyroxine (FT4) 0.759ng/dL (0.8–1.9), follicle-stimulating hormone (FSH) 90.1IU/L (postmenopause 21.7–153), luteinizing hormone (LH) 35.6IU/L (postmenopause 11.3–39.8), insulin-like growth factor (IGF-1) 258μg/L (81–225), somatotropin (GH) 5.75μg/L (0.06–5), corticotropin (ACTH) 21.3μg/L (10–46), cortisol 12.9μg/dL (5–25), prolactin (PRL) 9.95μg/L (4.6–37). A 75-g oral glucose tolerance test (OGTT) was performed (Table 1).
Acromegaly secondary to pituitary macroadenoma was diagnosed, and the study was completed by a colonoscopy that detected several tubulovillous adenomatous polyps, two of them with adenocarcinoma foci, and several serrated adenomas. Laparoscopic sigmoid resection was initially performed, and transsphenoidal surgery was subsequently performed on the pituitary macroadenoma.
A pathological examination found that most of the sample consisted of normal pituitary gland, while one third corresponded to a homogeneous tumor whose cells stained for GH only.
Postoperative MRI showed no tumor remnants. However, increased IGF-1 levels persisted (416) and the OGTT was pathological. Treatment was therefore started with somatostatin analogs. IGF-1 levels normalized at subsequent visits. Screening for hypercortisolism was negative, and thyroid ultrasound revealed several subcentimetric nodules without suspicious radiographic data.
PRKAR1A is a key component of the cAMP cell signaling pathway involved in tumorigenesis. CC may be considered as a form of multiple endocrine neoplasia with the involvement of the adrenal glands, the pituitary gland, the thyroid gland, and the gonads.
Clinical diagnosis is based on the presence of two or more typical signs confirmed by histology, laboratory tests, or imaging tests. Signs include skin lentiginosis with a typical distribution (lips, conjunctiva, oral and genital mucosa), blue nevus, psammomatous melanotic schwannoma, skin or mucosal, breast or cardiac myxomas, pigmented micronodular adrenal hyperplasia (PPNAD) acromegaly, thyroid carcinoma or multiple hypoechoic thyroid nodules, large cell calcifying Sertoli cell tumor, osteochondromyxoma and/or multiple ductal breast adenomas. If the patient is known to carry a mutation or is a first degree relative of an affected individual, only one sign is required for diagnosis.2 The genetic study should be proposed to all index cases and if a mutation is detected, also to first degree relatives.3 The most common and specific endocrine manifestation is PPNAD. Cardiac myxomas are the most common cause of mortality, either directly or as the result of surgical complications. They may be multiple and occur at an earlier age, and have a greater risk of recurrence as compared to sporadic ones.4,5
In the initial description of 1985, 4 out of 40 patients with CC had GH-secreting adenomas.4 More recent series report a 12% prevalence. However, a greater proportion of patients have changes in the axis occurring as baseline GH/IGF-1 elevation or an absence of control in OGTT.6,7 Acromegaly in CC has a slow, insidious course. Mean age at presentation is 35 years, and the condition usually occurs as a macroadenoma.1
Unlike patients with CC, mice heterozygous for mutations in PRKAR1A do not develop pituitary tumors, although GH-secreting tumors occur in knockout mice for this gene in the pituitary Pit1 line. Based on these animal models, it has been postulated that the complete loss of the healthy allele is required for the development of GH-secreting adenomas in CC. No mutations in the PRKAR1A gene have been detected in sporadic cases, but some data suggest that a non-genomic mechanism may cause the loss of expression of this protein, contributing to its development.8
Histopathologically, adenomas in CC may be similar to sporadic adenomas, be multifocal or have foci of hyperplasia of somatotroph cells. The presence of generalized hyperplasia of somatotroph cells in autopsies of patients with CC provides an explanation for the high prevalence of biochemical changes in the somatotropic axis in these patients.9 Unlike in our case, most stain for several hormones, mainly PRL, but staining for subunit-α, TSH-β, LH-β and, occasionally, FSH-β is also possible.
Seventy-two percent of patients concurrently have acromegaly and PPNAD.9 PPNAD was not detected in our case.
Our patient met clinical diagnostic criteria, and genetic study subsequently confirmed a mutation in the PRKAR1A gene. Her children do not currently meet the clinical diagnostic criteria and are pending the genetic study.
Please cite this article as: Rojo Álvaro J, et al. Acromegalia en paciente afecto de complejo de Carney. Endocrinol Nutr. 2013;60:277–8.
