







Pregnancy results in a significant change in both pituitary gland size and function. Due to this physiological adaptation, the diagnosis and management of pituitary diseases during pregnancy represents a particularly complex challenge. The presence of a functioning pituitary adenoma may be harmful to the health of the mother and fetus, and scientific evidence regarding the safety of drugs normally used to control hormone excess during pregnancy is scarce. In addition, pregnancy may be associated with the risk of the growth of a pre-existing pituitary adenoma. This review focuses on the diagnostic challenges in pregnant women with adenomas secreting prolactin, growth hormone, or adrenocorticotropic hormone. Some evidence-based recommendations for the treatment of these conditions during pregnancy are provided, and algorithms that could help monitor a pituitary adenoma during pregnancy are examined. Mention is also made of how hormone replacement therapy can be optimised in pregnant women with hypopituitarism. Finally, differential diagnosis between Sheehan's syndrome and lymphocytic hypophysitis, two pituitary disorders that may occur during pregnancy or delivery, is discussed.
La gestación determina un cambio significativo tanto del tamaño como de la función de la hipófisis. Como consecuencia de esta adaptación fisiológica, el diagnóstico y el manejo de las enfermedades hipofisarias durante el embarazo representan un reto particularmente complejo. La presencia de un adenoma hipofisario funcionante podría ser perjudicial para la salud de la madre y del feto, y la evidencia científica sobre la seguridad durante el embarazo de los medicamentos normalmente utilizados para controlar el exceso hormonal es escasa. Además, el embarazo podría estar asociado con el riesgo de crecimiento de un adenoma hipofisario preexistente. Esta revisión se centrará en las dificultades de diagnóstico en mujeres embarazadas portadoras de adenomas secretores de prolactina, hormona de crecimiento o adrenocorticotropina. Se proporcionarán algunas recomendaciones basadas en la evidencia para el tratamiento de estas condiciones durante la gestación y presentaremos algunos algoritmos que podrían ayudar a realizar el seguimiento de un adenoma hipofisario a lo largo del embarazo. Además, se hará mención de cómo optimizar la terapia hormonal sustitutiva en gestantes con hipopituitarismo y se ilustrará el diagnóstico diferencial entre el síndrome de Sheehan y la hipofisitis linfocitaria, dos trastornos hipofisarios que podrían ocurrir durante el embarazo o el parto.
Pituitary function undergoes significant changes during pregnancy due to physiological production of hormones by the placenta. The pituitary gland grows in height approximately 0.08 mm per week, and at the end of pregnancy its volume increases by up to 136% (45% in the first trimester) as compared to the previous size.1 It should be noted that pituitary growth during pregnancy is mainly due to lactotroph cell hyperplasia.
Such increase in the size of the pituitary gland may hinder interpretation of biochemical and imaging tests. New-onset or pre-existing pituitary disease during pregnancy is a challenge for endocrinologists, not only because of the difficulties to recognize and diagnose it, but also because of the scant scientific evidence on the most adequate treatment options. Few specific recommendations are available in the literature, and many of them are based on the approach established in non-pregnant women. Because of the potential risk that these conditions represent for both the mother and the fetus, it is essential to monitor them closely and treat them quickly and successfully, ensuring patients a multidisciplinary approach that involves, in addition to the endocrinologist, the gynecologist, the radiologist, and the neurosurgeon.
HyperprolactinemiaChanges in the hypothalamic-pituitary-lactotropic axis during pregnancyThe stimulant action of estrogens of placental origin on prolactin (PRL) synthesis and proliferation of pituitary lactotroph cells was described in 1978 by Lieberman.2 Subsequent studies confirmed that pituitary volume starts to increase in the first weeks of pregnancy until it reaches an average height of 8.8 mm in the third trimester, with 9.6−10 mm being the upper limit of the normal range.3 Maximum pituitary volume is reached during the first three days after delivery, when gland height may be up to 10.2–12 mm.3 PRL levels increase during pregnancy, in parallel to the increase in estrogen levels.4 Progesterone also stimulates production of PRL, which responds to the same factors that stimulate PRL secretion in non-pregnant women, including thyroid-stimulating hormone (TRH), sleep or food intake.5 During pregnancy, PRL increases from a mean concentration of 28.8 ng/mL (16.3–57.6 ng/mL) in the first trimester to a concentration of 216 ng/mL (124−318 ng/mL) a few weeks before delivery.4 The etiology of macroprolactin during pregnancy is heterogeneous. Although the big big prolactin usually accounts for 10%–30% of total PRL, macroprolactinemia associated to prolactin autoantibodies is found in 2.9 %–3.8% of pregnant women.6 Pituitary volume returns to normal within six months after delivery.3
Influence of pregnancy on prolactinomasProlactinomas account for 40% of all pituitary adenomas and are the most common pituitary neoplasm in women of childbearing age.7
Women with untreated prolactinoma cannot become pregnant because hyperprolactinemia affects pulsatility of the gonadotropin-releasing hormone (GnRH), decreases FSH and LH secretion, and interferes with formation of the corpus luteum and with progesterone secretion, inducing amenorrhea, infertility, and hypogonadism.8 As a result, diagnosis of prolactinoma is made in most cases before conception. Dopamine agonists (DAs), which are the first choice treatment option in prolactinomas, normalize PRL levels in 80 %–90% of cases and allow for resumption of the menstrual cycle.9 More than 80% of patients achieve pregnancy after successful management of hyperprolactinemia with Das.9 It should be emphasized that in patients treated with DAs, ovulation is restored before PRL levels normalize and pregnancy could therefore occur a few weeks after the start of treatment. It is important to inform the patient about this risk and to advise her to take mechanical contraceptive measures.9
Adenoma growthProlactinomas have estrogen receptors and can therefore grow during pregnancy.9 A potential increase in volume is also related to the initial size of the neoplasm.1–10 The risk of volumetric increase of a microprolactinoma is 1.5%–4.5%, while symptomatic growth, associated with headache and/or visual disturbances, occurs in 2% of cases. Because of the low risk of clinically relevant increase, discontinuation of treatment with DAs when pregnancy is confirmed appears to be a safe practice.11
In macroprolactinomas, the probability of symptomatic growth is greater than in microadenomas, and is 20%–30% in patients who have received no prior surgery or radiotherapy. In patients pretreated, the risk is 5%.11 It is therefore advisable to plan pregnancy after treatment has significantly reduced adenoma size, confining it within the sella turcica, especially if the lesion was located close to the optic chiasm.12
It should be noted that structural changes occurring in the pituitary gland during pregnancy may hinder interpretation of magnetic resonance imaging (MRI). In particular, an increased intensity is seen in the T1 sequence that, on one hand, may be misinterpreted as bleeding and, on the other hand, makes it more difficult to distinguish between the anterior and posterior pituitary gland.1
PRL levelsThe value of serial measurement of PRL during pregnancy is controversial. It could probably only be useful in patients with macroprolactinoma, because levels above the specific limit established for each gestational trimester would suggest potential adenoma growth.13 However, although some authors suggest that if PRL levels increase to reach those seen at diagnosis, it could be advisable to perform MRI without contrast medium, the more robust recommendations do not support prolactin measurement during pregnancy because of its poor reliability.12–15
Treatment of prolactinomas when pregnancy is plannedIf pregnancy is desired, continued treatment with DAs for at least one year before conception should be recommended to maximize the probability of a significant reduction in adenoma volume, which could in turn decrease the potential risk of symptomatic adenoma growth during pregnancy.10 Surgery may be a valid treatment option for both DA-resistant women and those who achieved no significant reduction in adenoma size despite normalization of prolactin levels.12 Transsphenoidal surgery does not appear to alter gonadal function in most cases but, as is well known, success of surgery mainly depends on the degree of experience of the neurosurgeon.14
As soon as pregnancy is confirmed, discontinuation of treatment with DAs, which cross the maternal-fetal barrier, is recommended to minimize fetal exposure to these molecules during the crucial period of organogenesis, also considering that the probability of tumor growth in the first trimester is low.12
Of the DAs available, cabergoline, quinagolide and bromocriptine, more safety studies have been published for the latter, which is therefore considered as the first choice. Treatment with bromocriptine appears safe, as the rates of spontaneous abortions, fetal malformations, and postnatal developmental changes in treated women are similar to those reported in healthy women.9 The fetal malformation rate in 6000 pregnancies exposed to bromocriptine in the first weeks of pregnancy was 1.8%, as compared to 6% in healthy women.16 Moreover, no increase in teratogenicity was documented in 100 women treated with bromocriptine throughout pregnancy.11,16 Cabergoline also appears to be safe, although there is less published data. The long half-life of cabergoline (79−115 h in hyperprolactinemic patients) causes the fetus to be exposed to the drug for many weeks despite its rapid discontinuation when pregnancy is confirmed.17 Although the total rate of maternal-fetal complications was not different in pregnant women taking cabergoline from that found in untreated women, two cases both of epilepsy and generalized developmental disorder were documented in a series of 61 children who had been exposed to cabergoline during the fetal period.17 While quinagolide appears to be safe and its use may represent an advantage during pregnancy because of its short half-life (22 h), it is not usually used frequently.18
TreatmentFig. 1 shows an algorithm for the management of prolactinomas during pregnancy.

At least two months before a planned pregnancy, it would be appropriate to replace cabergoline with bromocriptine. Since the tolerability profile of bromocriptine could be lower than that of cabergoline, bromocriptine should be started at a dose of 1.25–2.5 mg once daily for 3–7 days to decrease the chance that the patient experiences orthostatic hypotension and nausea. The dose may be gradually increased, every 2–3 days, to 2.5 mg twice daily. Subsequent treatment adjustments should be made after prolactin measurement and clinical re-evaluation of the patient.11
When faced with a treatment-resistant extrasellar macroadenoma, surgery may be advisable before planning pregnancy, but treatment should be individualized due to the lack of incontrovertible data on the best treatment option.15 The clinician should provide the patient with clear and objective information on the treatments available and accompany her throughout the decision-making process.9
Treatment with bromocriptine should be discontinued when pregnancy is confirmed, in both microprolactinomas and macroprolactinomas, with regression documented after surgery or long-term, successful treatment with DAs.9
In the presence of macroadenomas that have been inadequately treated before conception, or extrasellar masses that compress the chiasm (with a normal visual field), continued treatment may be indicated with DAs, particularly bromocriptine, since the safety data related to this option are limited.1,9,14
The possible occurrence of symptoms suggesting tumor growth (headache, visual impairment) should be assessed every month in macroadenomas and every three months in microadenomas. Visual field examination should be performed every three months in macroadenomas, or even earlier if compression symptoms appear or if the tumor is close to the optic chiasm.15 In this case, a non-contrast MRI should be performed and, if tumor growth is documented, treatment with bromocriptine should be started. If vision remains impaired (or even worsens) after three weeks of treatment, surgery in the second trimester is a recommended option.15
Two months after delivery, prolactin measurement and a repeat MRI should be performed.
Breastfeeding is permitted in women who do not take DAs and who have a stable microadenoma or macroadenoma successfully treated before conception. In patients with a macroadenoma that has grown or required treatment with DAs during pregnancy, breastfeeding is not advised.9
Postpartum remission of a microprolactinoma with persistent normalization of prolactin levels has been documented in up to 35% of patients due to ischemia/necrosis or regressive transformation of the adenoma during pregnancy.9
AcromegalyChanges in the hypothalamic-pituitary-somatotropic axis during pregnancyIn the first trimester of physiological pregnancy, the rapid increase in estrogen levels decreases the levels of insulin-like growth factor-1 (IGF-I), while a compensatory increase in pituitary growth hormone (GH) levels has not been reported in all studies. From the fifth week of pregnancy, placental GH (GH-V) production begins, and increases gradually until a plateau is reached after 30 weeks.19 GH-V secretion is continuous and not controlled by the GH-releasing hormone (GHRH) or ghrelin. In healthy women, GH-V stimulates IGF-I production in the liver with the resultant inhibition of pituitary GH.19 In the last weeks of pregnancy, GH-V levels range from 13 and 25 μg/L, depending on the test method used, with an interindividual coefficient of variation ranging from 10 and 60 μg/L. IGF-I levels at the end of pregnancy almost double the pregestational values and depend on the balance between the stimulating effect of GH-V, the concentrations of which could be comparable to those of pituitary GH in active acromegaly, and hepatic resistance to GH induced by hyperestrogenism.20
In women with acromegaly, the somatotropic adenomatous cells do not respond to IGF-I inhibition, and pituitary GH levels therefore remain elevated throughout pregnancy.21 However, hyperestrogenism associated to pregnancy induces hepatic resistance to GH, preventing excessive increases in IGF-I levels in the first trimester in both women with acromegaly and healthy women, women with type 1 diabetes, and those with hypopituitarism treated with GH.22 In fact, in pregnant acromegalic patients, IGF-I levels significantly decrease during the first trimester as compared to preconception values, and could be associated to a subjective improvement in the typical symptoms of the disease.23,24 In addition, women with a macroadenoma or those who received treatment with somatostatin analogues (SAs) or DAs before pregnancy more frequently had IGF-I levels that were stable or reduced by more than 30% at any time during pregnancy as compared to a microadenoma or no previous treatment respectively.23 Although IGF-I reduction in pregnant women with acromegaly may partially depend on a carry-over effect of prior treatment, the inhibitory effect of estrogens appears to be prevalent.23 It should be noted, however, that IGF-I levels may increase very significantly in the second half of pregnancy as a result of concomitant elevation of pituitary GH and GH-V.21
DiagnosisMost cases of pregnancy reported in women with acromegaly occurred in patients who had already achieved disease control. Active acromegaly is related to gonadal dysfunction due, on the one hand, to the harmful effect of excess GH/IGF-I on the gonadotropic axis and the ovary, and on the other hand, to hyperprolactinemia (documented in one third of patients) and hypogonadotropic hypogonadism, caused by the “mass effect” of the adenoma.25,26
Diagnosis of acromegaly during pregnancy is challenging. The normal ranges commonly used for GH measurement, both in basal conditions and after oral glucose overload, cannot be applied in pregnant women because pituitary GH decreases and IGF-I increases physiologically in pregnancy. Therefore, diagnosis of acromegaly should be delayed, whenever possible, until after delivery. However, high IGF-I levels in the first trimester or GH levels, detected by ultrasensitive methods, above 1 μg/l in the third trimester may suggest acromegaly.21 Pituitary MRI to confirm the diagnosis of acromegaly should be performed from the second trimester only if strictly necessary, i.e. if there are compression symptoms associated with the probable presence of a macroadenoma.
It should be noted that the most common immunoenzymatic methods do not recognize GH-V due to the high cross-reactivity between antibodies and isoforms of GH. The placental variant cannot therefore be differentiated from pituitary GH. Diagnosis of acromegaly during pregnancy is based on the use of tests with two monoclonal antibodies, one of which is specific for pituitary GH and the other for GH-V.21
Maternal-fetal complicationsThere is little evidence on maternal-fetal complications in acromegaly, and the few data reported in the literature come from studies including small patient samples.
Worsening of acromegaly symptoms was reported in four of 24 patients (17%), one of whom opted for therapeutic abortion in week 10 due to severe symptom exacerbation.20 Recurrence of GH hypersecretion and return of clinical signs of acromegaly were documented in a patient with macroadenoma in whom treatment with bromocriptine was discontinued during pregnancy.27 Another patient showed signs of increased intracranial pressure due to adenoma re-expansion in week 39 of pregnancy.28
Metabolic and cardiovascular complications of acromegaly may represent a risk for both the mother and the foetus. A trend to a greater prevalence of hypertension and gestational diabetes mellitus (GDM) was seen in 46 pregnant women with acromegaly as compared to the general French population, with no difference between patients with and without control of GH/IGF-I hypersecretion.24 The risk of developing glucose intolerance or GDM was 60% and 13 %–32% respectively in pregnant women with acromegaly, a condition which is in itself associated to insulin resistance.24 It should be noted that GDM occurred in a patient who had been treated with SAs during the first half of pregnancy, suggesting that this complication can be seen regardless of medical treatment.24 In any case, cases of GDM reported to date have been mild and have been controlled with specific dietary regimens alone.20 Similarly, hypertension, reported in 13.6% of pregnant women with acromegaly, has not been associated with pre-eclampsia or any other maternal complication.20 Prospective studies would be needed to establish the effective prevalence of these complications in acromegalic women.
Most GH-secreting tumors are macroadenomas and frequently show prolactin co-secretion. All of this, combined with the physiological increase in size of the pituitary gland during pregnancy, could potentially induce adenoma progression or promote the occurrence of tumor infarction in pregnant women with acromegaly.20 This potential risk increases with initial tumor size, and is minimal in microadenomas. In most patients who had been treated with surgery or radiotherapy before becoming pregnant, no clinically relevant growth of the residual tumor was documented, which suggests that treatment should be optimized before planning pregnancy.20 A retrospective study of 59 pregnancies in 46 women with acromegaly (in six of whom it was diagnosed during pregnancy) reported that only four women were found visual field defects, which led to the diagnosis of acromegaly in three of them.24 The other patient, who had a GH- and PRL-secreting macroadenoma and was being treated with bromocriptine, developed intratumoral hemorrhage at week 34.24 In the six months following delivery, no increase was found in adenoma volume (macroadenoma in most cases) in 22 of 27 (81%) patients with available MRI. An enlargement was found in 11% of patients with macroadenoma, one of whom had previously been treated with surgery and bromocriptine.24 Headache was recorded in seven patients, and one of them, who had a stable MRI at 22 weeks of pregnancy, developed diplopia three months after delivery.24
Although some cases of spontaneous abortion and fetal loss have been reported, no malformation has been documented in newborns of women with acromegaly, and weight is usually normal in most of them, which is consistent with placental impermeability to GH and IGF-I29. Lactation is not associated with tumor growth and is therefore usually not contraindicated, especially in women with minimal residual tumor or with no apparent residues.24,25
TreatmentFig. 2 shows an algorithm for the management of pregnant women with acromegaly.

Scheduling of pregnancy and normalization of GH and IGF-I levels before conception are recommended. In addition, SAs should be discontinued at least two months before a scheduled pregnancy. The safety data on SAs during pregnancy reported in the past 15 years are usually encouraging, and until their continued use during pregnancy they have not been associated with any maternal complications or fetal malformation in the few, rather anecdotal, studies published to date.30,31 However, use of these drugs during pregnancy should be avoided because of the lack of comprehensive information about their safety profile.32 In fact, all five somatostatin receptor subtypes are expressed in the placenta, and octreotide crosses the placental barrier.33 Fetal intrauterine growth retardation was documented in a woman treated with long-acting octreotide (LAR) throughout pregnancy, and a larger series reported that women treated with SAs during pregnancy were more likely to have microsomic newborns.23,31 This may be partially explained by the reduction in the uterine artery systolic flow velocity and the systolic velocity seen in a pregnant woman with acromegaly after octreotide injection.33 Lanreotide or pasireotide are in category C of the Food and Drug Administration (FDA) due to the evidence of fetal complications in animal models.34 Use of pegvisomant in 35 pregnancies (with maternal and paternal drug exposure in 27 and eight cases respectively) was recently reported. Fourteen of 18 live newborns were normal, while data on the remaining four were not reported.35 Pegvisomant levels in fetal circulation appear to be minimal, suggesting a low or absent transplacental passage.36 Dopamine analogues may be associated with a high risk of macrosomy that should be confirmed in future studies.23
During pregnancy, hormone levels need not be controlled, but blood pressure and blood glucose should be strictly monitored, as well as symptoms suggesting possible tumor growth (blurred vision, headache). Visual field examination should be performed every three months, or even earlier if chiasmal compression is clinically suspected or when the tumor is located close to the chiasm.20 Treatment may be started with SAs or bromocriptine if there is headache, and MRI could be performed from the second trimester. If visual impairment is found, surgery is indispensable.28
A cesarean section may be advisable in the presence of macroadenoma because of the risk of pituitary apoplexy during delivery.28
Breastfeeding is not permitted in patients on medical treatment. Women who have undergone surgery for a macroadenoma and have minimal or no residues may breastfeed, as well as women with microadenomas.29
Cushing’s diseaseChanges in the hypothalamic-pituitary-adrenal axis during pregnancyPregnancy is characterized by physiological hyperactivation of the hypothalamic-pituitary-adrenal (HPA) axis. The placental production of both corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) increases exponentially during the first trimester.37 However, the concomitant increase in CRH binding protein protects the HPA axis from excessive CRH exposure until the end of the second trimester. In the third trimester, bioavailable CRH increases again, because it plays a significant role in fetal lung maturation and delivery.38
As a result of CRH/ACTH hypersecretion, a 1.6-fold increase is seen in circulating cortisol levels from week 11 of pregnancy. Although cortisol secretion maintains the circadian variations, continues to increase during pregnancy.39 In the third trimester, urinary free cortisol (UFC) levels are three-fold higher than pre-pregnancy levels. However, the placenta also expresses the enzyme 11β hydroxysteroid dehydrogenase 2 (11β HSD2) that converts cortisol to cortisone, protecting the fetus from maternal hypercortisolism.39
Fetal adrenal glands are larger than those in adults and produce both dehydroepiandrosterone (DHEAS) and cortisol. Two-thirds of the cortisol found in the fetal circulation come from the fetal adrenal glands, while the remaining third is of maternal origin. In addition, fetal cortisol is derived on the one hand from placental progesterone conversion, and on the other hand from cortisone conversion in the choriodecidual space.40
Estrogens also induce an increase in corticosteroid binding globulin (CBG), which reaches peak levels at the end of pregnancy, resulting in overestimation of serum cortisol measured by immunoenzymatic methods.41
Approximately four days after delivery, CRH, ACTH, and cortisol levels gradually decrease toward pre-pregnancy levels. It should be noted that a certain degree of suppression of CRH secretion occurs during the first three months after delivery. This may contribute to the emotional disorders and autoimmune diseases commonly seen in this period.40
DiagnosisPregnancy is uncommon in patients with Cushing’s syndrome (CS) due to associated fertility problems such as oligo/amenorrhea and hypogonadotropic hypogonadism. The adrenal cause appears to be most common in pregnant patients, in contrast to what is seen outside the gestational period. A review of cases reported between 1952 and 2015 showed that 44% of women had CS of adrenal origin, while 28% had pituitary Cushing’s disease (CD).42
The signs and symptoms associated to hypercortisolism resemble those that may appear during pregnancy, including weight gain, striae, hypertension, and GDM, hindering differential diagnosis between Cushing syndrome and pregnancy. In any case, presence of features suggesting a catabolic state such as muscle weakness, wide purple striae, especially extraabdominal, and osteoporosis supports the suspicion of pathological hypercortisolism.41
Diagnostic confirmation based on hormone measurements is also very difficult because in normal pregnancy there may be an increase in serum and urinary cortisol levels, as well as an incomplete response to dexamethasone suppression (Table 1).
Value of diagnostic tests in Cushing’s syndrome (CS) in pregnancy.
| Test | Pregnancy | Value in CS diagnosis |
|---|---|---|
| Physiological | ||
| UFC | Up to 3-fold increase in 2nd & 3rd trimesters | Only if an increase greater than 3 times the range is found |
| 1-mg DST | Suppression attenuated | Low |
| Cortisol in saliva/blood at midnight | Increased but with preserved rhythm | Useful if absence of rhythm is documented, but there are no validated ranges for pregnancy |
| ACTH | Increased | Unhelpful, may not be suppressed in AR adenomas |
| CRH test | Reduced response | Helpful (FDA category C) |
| Desmopressin test | Reduced response | Yes (80% sensitivity) |
| HDDST (2 mg every 6 h for 2 days)/8-mg DST (overnight) | Suppression attenuated | Yes |
| >50% suppression in CD vs ectopic CS | ||
| >80% suppression in CD vs adrenal CS | ||
| Pituitary MRI | – | Scarce; without gadolinium does not identify microadenomas |
| IPSC | – | Avoid |
1-mg DST: 1-mg dexamethasone suppression test; ACTH: adrenocorticotropic hormone; UFC: 24-h urinary free cortisol; CRH: corticotropin-releasing hormone; IPSC: inferior petrosal sinus catheterization; DST: dexamethasone suppression test; CD: Cushing’s disease; FDA: Food and Drug Administration; HDDST: high-dose dexamethasone suppression test; MRI, magnetic resonance imaging; CS, Cushing’s syndrome; AR, adrenal.
Assessment of cortisol circadian rhythm may be considered the cardinal test because, as previously described, cortisol circadian rhythm remains intact in physiological pregnancy. Absence of a midnight nadir in cortisol secretion, assessed in saliva or serum, is highly suggestive of CS, especially if there is a concomitant elevation of UFC levels to more than three times the upper limit of the normal range.41 ACTH measurement cannot be used to differentiate adrenal CS from CD, because pituitary secretion of ACTH is stimulated by placental CRH, and serum levels will therefore remain elevated even in adrenal CS. ACTH levels in pregnant women with CD are in the upper half of the normal range or even above the same cut-off point.43 The high-dose dexamethasone suppression test (HDDST) may be helpful for differentiating CD from ACTH-independent CS, as cortisol levels >80% of basal level suggest a pituitary origin.39 The high levels of cortisol bound to CBG seen during pregnancy could be associated to a lack of suppression, even in the case of CD.44 Alternatively, the desmopressin or CRH tests may be used, although CRH is in category C of the Food and Drug Administration (FDA), and its use is therefore recommended only when there is a clear clinical indication.37
Inferior petrosal sinus catheterization (IPSC) is not recommended due to the use of ionizing radiation and the risk of thrombotic events. In any case, use of IPSC has been reported in some isolated cases in which the direct jugular approach was chosen to minimize fetal exposure to radiation.43
MRI should be performed without gadolinium from the second trimester and only if strictly necessary, that is, when the diagnosis is true and surgery is planned, but may not disclose a very small microadenoma.43 In addition, pituitary gland enlargement during pregnancy could mask smaller tumors.42 Abdominal CT should of course be avoided, while abdominal ultrasonography is very helpful because of the higher prevalence of CS of adrenal origin as compared to pituitary origin.41
Maternal-fetal complicationsCS in pregnancy is a serious condition that must always be treated due to the high incidence of life-threatening complications in both the mother and the fetus. The prevalence rates of hypertension and diabetes in pregnant women with CS are 68% and 25% respectively.41 The risk of pre-eclampsia is six times higher in these women than in healthy pregnant women, and the risks of spontaneous abortion and fetal loss are 2 and 10 times higher respectively.41 Mortality associated to pregnancy is significantly higher in women with CS than in healthy women, and even higher than the mortality reported in southern Sudan, the region of the world with the highest mortality rate in pregnant women (mortality rate in healthy women worldwide, 209/100,000 live births; in Sudan, 956.8/100,000; in CS, 1257/100,000).41 In addition, patients with active CS have a significantly higher risk of hypertension, GDM, pre-eclampsia, and fetal loss as compared to women with CS in remission. It should be noted that in the latter patients, fetal mortality is similar to that seen in healthy women, which emphasizes the fact that control of hypercortisolism is essential to maximize the chance of a successful pregnancy. However, biochemical remission of CS does not decrease the risk of prematurity.41
TreatmentTranssphenoidal surgery is the treatment of first choice and should be performed in the second trimester.41 Bilateral adrenalectomy is the last resort for management of persistent and/or progressive disease.42 In a retrospective analysis of 136 pregnant women with CS, unilateral or bilateral adrenalectomy performed in 31 patients was associated to a live birth rate of 87%.45
There are limited data on the use of medical treatment in pregnancy. Metyrapone, the most commonly used drug, controls hypercortisolism in most cases and is usually well tolerated. Although metyrapone crosses the placental barrier, no fetal abnormalities attributable to its use during pregnancy have been reported to date.46 However, women treated with this drug should be closely monitored, because metyrapone induces an increase in mineralocorticoid precursors, such as 11-deoxycorticosterone, that could worsen hypertension and promote the occurrence of pre-eclampsia.46,47 Treatment with cabergoline could be an option in pregnant women with CD, taking into account its limited efficacy and its potential in non-pregnant patients.48 Although ketoconazole was not associated to any incidence in the few cases reported, its use in pregnancy is not advised due to the anti-androgen effect and potential teratogenicity (FDA category C)46. Use of aminoglutethimide and mitotane is contraindicated during pregnancy.46
Non-functioning adenomas and other sellar region lesionsSurgical resection is advised before conception in all patients with non-functioning macroadenomas (NFMAs) who want to become pregnant, particularly if they have growing tumors close to the chiasm, or when a chiasmal syndrome already exists. The risk of pituitary apoplexy during pregnancy or after delivery should also be considered.49
NFMAs do not usually increase in size during pregnancy and are not associated with maternal-fetal complications.49 However, physiological enlargement of the pituitary gland may cause hormonal deficiencies, headache, and visual disturbances due to compression of the optic nerve (or, more rarely, the external ocular motor nerve). All non-functioning sellar lesions should be monitored for potential growth symptoms and visual field assessment (for masses >1 cm) every three months.49
In the presence of compressive symptoms or NFMAs close to the chiasm, a monthly visual field examination is recommended. If growth is documented, surgery is recommended in the second trimester.1 If surgery is contraindicated, treatment could be started with cabergoline (mean dose, 1.5 mg/week; maximum dose, 3.5 mg/week), but this approach is not supported by much evidence.50 If visual impairment worsens, this treatment should be discontinued.
In addition to pituitary adenomas, other tumor (craniopharyngiomas, meningiomas), cystic (Rathke cyst, arachnoid cyst), or vascular (aneurysms) lesions may develop in the sella turcica. As in pituitary adenomas, diagnosis should focus on measurement of basal pituitary hormones, and pregnancy should be planned after adequate neurosurgical assessment. Some studies suggested that meningiomas may grow during pregnancy due to increased vascular flow and/or cell proliferation. Resection of meningiomas before pregnancy is therefore advised.51
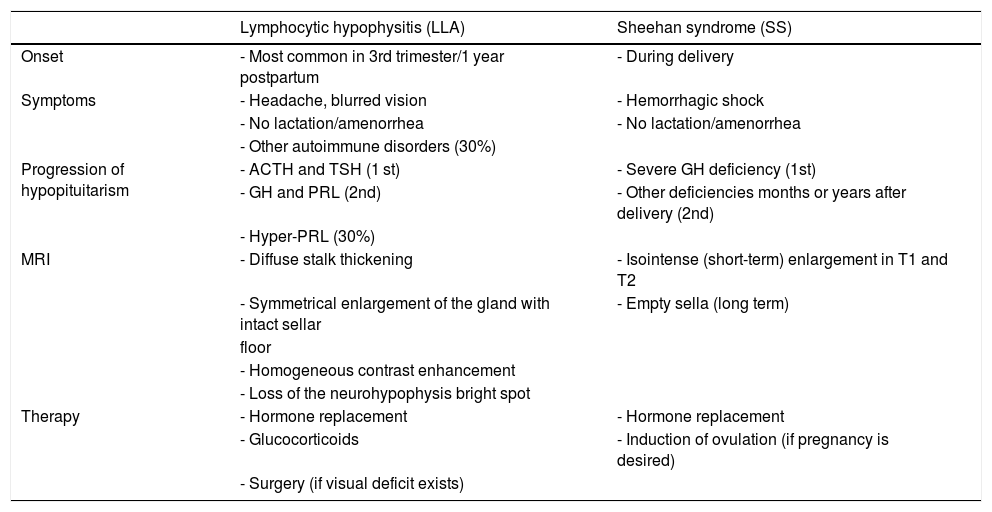
Lymphocytic hypophysitisLymphocytic hypophysitis (LH) is a rare autoimmune disorder characterized by infiltration of pituitary tissue by lymphocytes, plasmocytes, eosinophils, macrophages, and neutrophils, with the resultant fibrous degeneration of glandular parenchyma.52 LH, which is associated to other autoimmune disorders in 30% of cases, usually becomes manifest in the third trimester of pregnancy or in the first year after delivery, although some cases have been reported in the first months of pregnancy. LH is usually diagnosed in women who cannot breastfeed or who have persistent amenorrhea in the postpartum period.52 LH occurs as a sellar lesion that causes headache, visual disturbances, and hypopituitarism. ACTH and thyroid-stimulating hormone (TSH) deficiencies initially occur, later followed by hypoprolactinemia and hyposomatotropism. Hyperprolactinemia is seen in 30% of cases, while diabetes insipidus may occur if there is infundibular involvement53 (Table 2).
Differential diagnosis between lymphocytic hypophysitis (LH) and Sheehan syndrome (SS).
| Lymphocytic hypophysitis (LLA) | Sheehan syndrome (SS) | |
|---|---|---|
| Onset | - Most common in 3rd trimester/1 year postpartum | - During delivery |
| Symptoms | - Headache, blurred vision | - Hemorrhagic shock |
| - No lactation/amenorrhea | - No lactation/amenorrhea | |
| - Other autoimmune disorders (30%) | ||
| Progression of hypopituitarism | - ACTH and TSH (1 st) | - Severe GH deficiency (1st) |
| - GH and PRL (2nd) | - Other deficiencies months or years after delivery (2nd) | |
| - Hyper-PRL (30%) | ||
| MRI | - Diffuse stalk thickening | - Isointense (short-term) enlargement in T1 and T2 |
| - Symmetrical enlargement of the gland with intact sellar | - Empty sella (long term) | |
| floor | ||
| - Homogeneous contrast enhancement | ||
| - Loss of the neurohypophysis bright spot | ||
| Therapy | - Hormone replacement | - Hormone replacement |
| - Glucocorticoids | - Induction of ovulation (if pregnancy is desired) | |
| - Surgery (if visual deficit exists) |
ACTH: adrenocorticotropic hormone; GH: growth hormone; PRL: prolactin; MRI: magnetic resonance imaging; TSH: thyroid-stimulating hormone.
MRI is essential for diagnosis because it reveals a diffuse thickening of the stalk with symmetrical enlargement of the pituitary gland and an intact sellar floor, together with loss of the neurohypophysis bright spot in T1. Although these findings are typical of HL, half the patients are misdiagnosed with adenoma, which results in unnecessary surgery.53
The natural course of the disease varies and depends on the degree of aggressiveness, but both complete regression of the pituitary lesion and spontaneous, partial or complete, regression of hormonal dysfunctions have been reported.53
Seventy-three percent of patients affected require long-term replacement of one or more hormones.53
It should be noted that LH remains undiagnosed in a significant number of women, particularly when it occurs during pregnancy.54 LH should be suspected in pregnant women with hypopituitarism because early loss of corticotropic (and thyrotropic) function could threaten the life of the mother during or after delivery. Symptoms of hypoglycemia and hypothermia in the early postpartum period may also be suggest LH, and should therefore be recognized and treated quickly.55
In addition to hormone replacement therapy, glucocorticoids may be used if chiasmal compression and visual impairment occur. However, the efficacy of this treatment is limited and a high recurrence rate has been reported.56 Treatment with DAs may be useful in hyperprolactinemia because it can improve the visual field. Decompression surgery may be considered during pregnancy if glucocorticoids are ineffective, but if symptoms are mild, treatment should be delayed until after delivery.57
Sheehan syndromeSheehan syndrome (SS) is acute ischemic necrosis of the anterior pituitary gland due to massive intrapartum hemorrhage, with resultant hypovolemic shock. Although incidence of SS has decreased in developed countries thanks to improved obstetric care and advances in emergency medicine, its clinical presentation is heterogeneous and physicians should therefore be able to recognize the mildest forms.58
Manifestations of acute SS are rare and include headache, hypopituitarism, with early involvement of somatotropic cells, postpartum hyponatremia, coma, and death. In most patients, SS may cause vague symptoms such as anemia, weakness, and fatigue, while hormone deficiencies appear months to years after delivery, when partial or total empty sella syndrome has already developed.59 Late hypopituitarism may occur due to the effect of anti-hypothalamic and anti-pituitary antibodies, suggesting an autoimmune pathogenesis.1
In patients with acute SS, MRI shows pituitary enlargement that could be erroneously associated to LH. Differential diagnosis between these two conditions is important and is based on recognition of the symptoms and temporal progression of hormone deficiencies (Table 2). A clinical history positive for obstetric bleeding may reveal SS, but cases of HL during pregnancy complicated by postpartum bleeding have been reported.60 An additional condition that should be differentiated from SS is pituitary apoplexy, which occurs in 80% of cases in the presence of a pituitary macroadenoma, due to tumor expansion beyond the limits of its vascular supply and to changes in gland perfusion. Although pituitary apoplexy is commonly associated to severe symptoms, including severe headache, mental confusion, visual disturbances, and severe corticotropic deficiency, that require an emergency surgical solution, some patients only show hypopituitarism, amenable to replacement therapy until delivery.1
Treatment of SS consists of replacement of deficient hormones, while glucocorticoids are the life-saving measure in the acute phase. Women who wish another pregnancy should undergo ovulation induction, although some cases of spontaneous pregnancy have been reported due to late improvement of pituitary function.58
HypopituitarismHypopituitarism in pregnancy is associated to high risk of miscarriage, anemia, gestational hypertension, placental detachment, premature delivery, postpartum bleeding, and cerebrovascular accidents.61,62
Management of hypopituitarism in pregnancy is based on adequate hormone replacement according to patient needs in each trimester, with careful monitoring of potential overdoses. GH replacement does not appear to be necessary.63
ACTH deficiencyFunctional assessment of the pituitary-adrenal axis in pregnancy is difficult. Measurement of salivary cortisol in the morning is the test of first choice.64 To confirm adrenal insufficiency, the short stimulation test with ACTH 250 μg is performed. The cut-off point in the second and third trimesters is above 60%–80% as compared to healthy women. A cortisol peak above 30 μg/dL allows for ruling out adrenal insufficiency.64
Hydrocortisone is the glucocorticoid of choice, and it may have to be given at gradually increasing doses, particularly in the third trimester due to increased binding globulin levels. During delivery, patients should be given 50 mg of hydrocortisone as a bolus, followed by continuous infusion of 100−200 mg daily or 50 mg every six hours. The usual oral dose may be administered again two days after delivery. The newborn would have to be monitored for the potential occurrence of adrenal insufficiency, an extremely rare complication that was documented after intrauterine exposure to pharmacological doses of glucocorticoids, particularly dexamethasone.65,66 As mentioned above, the fetus is protected from maternal hypercortisolism by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) that converts cortisol to cortisone, metabolically inactive. Hydrocortisone replacement therapy is recommended in pregnancy because this glucocorticoid, unlike dexamethasone, is metabolized by 11β-HSD2.66
TSH deficiencyMonitoring of thyroxine treatment in pregnancy, because of the little value of TSH measurement, is based on testing throughout pregnancy of free T4 levels, that should remain in the upper limit of normal.67 From week 16, total T4 levels may be assessed, as measurement methods are less susceptible to interference from binding proteins and to hormonal changes typical of pregnancy. The normal range for total T4 should increase by 50% to reflect the actual values during pregnancy.67
Until week 12 of pregnancy, the mother is the only source of thyroid hormones for the fetus, and maternal hypothyroxinemia has been associated with cognitive delay in childhood. Dose of thyroxine should be increased 50% with monthly monitoring.67
Diabetes insipidusThe threshold for antidiuretic hormone (ADH) secretion and thirst perception are reduced by the effect of human chorionic gonadotropin (HCG).68
Transient diabetes insipidus (DI) may occur due to increased placental vasopressinase activity at the end of the second trimester and the beginning of the third trimester, particularly in twin pregnancies. Exacerbation of pre-existing DI is a rare event.68
Diagnosis is based on measurement of osmolality of sodium levels in serum and urine and monitoring of fluid intake/urine output. Treatment with desmopressin is safe, and is usually given at higher doses than in non-pregnant patients.69 A few days after delivery, desmopressin may be discontinued or, in the event of pre-existing DI, the pre-pregnancy dose may be given again.69
ConclusionsPituitary dysfunctions during pregnancy are rare, and their management is challenging not only because of the lack of scientific evidence in this field, but also because of the limited clinical experience. A multidisciplinary approach is essential to prevent maternal-fetal complications and to optimize treatment. Development of agreed protocols at each institution would be advisable, as well as the creation of records to help clarify the still controversial issues related to diagnosis and treatment, and to prepare guidelines shared at national and international level.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Valassi E. Patología hipofisaria y gestación. Endocrinol Diabetes Nutr. 2021;68:184–195.
