




Although X-linked hypophosphatemia (XLH) is regarded as a rare disease, it is the most common form of the heterogeneous group of hereditary disorders known as hypophosphatemic rickets. The prevalence of HLH is 1:20,000,1 and the origin of the disorder is a mutation of the PHEX gene, located on the short arm of chromosome 22, which encodes for a phosphate-regulating endopeptidase. Hundreds of mutational varieties have been described2 that encode for abnormal PHEX proteins, which lose their function of inhibiting fibroblast growth factor 23. This results in the suppression of the expression of the tubular cell sodium-phosphorus cotransporter, and thus produces a decrease in the tubular reabsorption of phosphate, with hypophosphatemia.3 In addition, fibroblast growth factor 23 suppresses the levels and activity of α-1-hydroxylase, leading to a decrease in the levels of 1,25-dihydroxyvitamin D, the active form of vitamin D.
The disease exhibits a broad phenotypic variability, and its signs and symptoms include: rickets, osteomalacia, short stature, lower limb bowing, craniosynostosis, bone pain, fractures, extraskeletal calcifications, dentinal defects and neurosensory deafness, among other manifestations. Studies have shown XLH to have a significant impact upon the quality of life of these patients, with reduced mobility and functional disability.4,5
The classical treatment of XLH has been based on the oral supplementation of phosphorus and calcitriol. Such treatment is associated with low patient adherence, due to poor gastrointestinal tolerance, and is not without significant adverse effects, such as nephrocalcinosis or secondary hyperparathyroidism.6 However, a new human monoclonal anti-fibroblast growth factor 23 antibody, burosumab, approved by the EMA and the FDA, and already in pediatric compassionate use in Spain, has opened up the possibility of blocking the mechanism of the disease and ensuring normal development in childhood.7 Furthermore, its possible role in adults is currently under study.8
The present letter reports two cases involving mutations not previously reported in the literature.
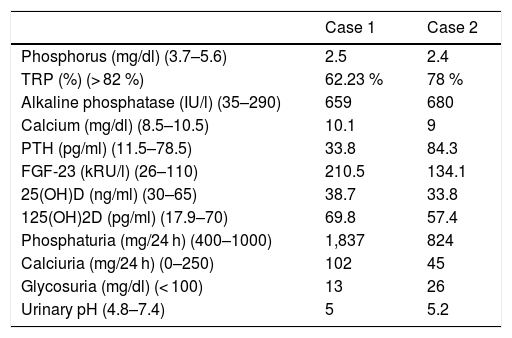
Clinical case 1A boy aged two years and 8 months was referred due to bilateral genu varum from the age of 9 months. Until then he had been followed-up on by Traumatology only; there was no other relevant medical history. Blood and urine tests were performed (Table 1), with findings consistent with XLH. Renal ultrasound showed no alterations. A genetic study was thus requested, and treatment with oral phosphorus and calcitriol was started.
Laboratory test findings at diagnosis.
| Case 1 | Case 2 | |
|---|---|---|
| Phosphorus (mg/dl) (3.7–5.6) | 2.5 | 2.4 |
| TRP (%) (> 82 %) | 62.23 % | 78 % |
| Alkaline phosphatase (IU/l) (35–290) | 659 | 680 |
| Calcium (mg/dl) (8.5–10.5) | 10.1 | 9 |
| PTH (pg/ml) (11.5–78.5) | 33.8 | 84.3 |
| FGF-23 (kRU/l) (26–110) | 210.5 | 134.1 |
| 25(OH)D (ng/ml) (30–65) | 38.7 | 33.8 |
| 125(OH)2D (pg/ml) (17.9–70) | 69.8 | 57.4 |
| Phosphaturia (mg/24 h) (400–1000) | 1,837 | 824 |
| Calciuria (mg/24 h) (0–250) | 102 | 45 |
| Glycosuria (mg/dl) (< 100) | 13 | 26 |
| Urinary pH (4.8–7.4) | 5 | 5.2 |
FGF-23: fibroblast growth factor 23; PTH: parathyroid hormone; TRP: tubular reabsorption of phosphate; 25(OH)D: 25-hydroxyvitamin D; 1,25(OH)2D: 1,25-hydroxyvitamin D.
The sequencing study of the PHEX gene identified the c.2182C > T; p.Gln728Ter mutation, a stop codon in apparent heterozygosis, despite being located on the X chromosome of a male. The patient was confirmed as having a single X chromosome (karyotype study [46 XY]) and a single allele of the PHEX gene (established using the multiplex ligation-dependent probe amplification [MLPA] method). The genetic study of the mother did not reveal the studied variant of the PHEX gene; the condition was therefore classified as a de novo mutation. To the best of our knowledge, this variant has not been previously reported in the literature. Apparent heterozygosis was identified in DNA from peripheral blood and saliva; it was therefore found in mesoderm and ectoderm, and demonstrated both by next-generation sequencing (NGS) and the Sanger method.
During follow-up, the patient required osteotomy and plaque placement to correct significant lower limb deformities.
Clinical case 2An 18-year-old woman had been diagnosed with XLH at two years and 7 months of age (Table 1). At that time it was already known that her mother, also affected by the disorder, was of extremely short stature and had marked lower limb bowing. In contrast, the patient suffered scant morbidity, with subsequent normal weight, height and skeletal development.
A genetic study was performed using the MLPA technique. The karyotype was characterized by a majority mosaic trisomy 47XXX (22/25 metaphases) as compared to 46XX, three copies of the PHEX gene being identified, except in exon 16, of which only two were detected. This finding, consistent with a deletion of that exon in one of the three copies, was also observed in the genetic study of the mother; this was therefore probably the cause of the disease. The fact that the patient presented X trisomy and that the deletion was in only one of the 3 copies could explain the scant clinical expressivity.
Since diagnosis, the patient has been treated with oral phosphorus and calcitriol, and although adherence proved erratic during adolescence, it is currently adequate. In these years she has developed secondary hyperparathyroidism and incipient nephrocalcinosis.
In conclusion, XLH is characterized by broad variability in its clinical expression, with no clear genotype-phenotype relationship, and management of the disease requires a multidisciplinary team of specialists. Moreover, it should be kept in mind that there are scantly symptomatic forms of the disease that are difficult to diagnose in the absence of affected relatives, particularly when they manifest in adulthood with chronic bone pain, fractures or pseudo-fractures, early osteoarthritis, dental problems, or enthesopathy.
Financial supportNo funding has been received for the present study.
Conflicts of interestThe authors state that they have no conflicts of interest related to this paper.
Please cite this article as: Broseta JJ, López-Romero LC, Cerón JA, Mendizábal S, Hernández-Jaras J. Mosaicismo en 2 casos de hipofosfatemia ligada al cromosoma X. Endocrinol Diabetes Nutr. 2020;67:70–71.
