





The irruption of lipoprotein(a) (Lp(a)) in the study of cardiovascular risk factors is perhaps, together with the discovery and use of proprotein convertase subtilisin/kexin type 9 (PCSK9i) inhibitor drugs, the greatest novelty in the field for decades. Lp(a) concentration (especially very high levels) has an undeniable association with certain cardiovascular complications, such as atherosclerotic vascular disease (AVD) and aortic stenosis. However, there are several current limitations to both establishing epidemiological associations and specific pharmacological treatment. Firstly, the measurement of Lp(a) is highly dependent on the test used, mainly because of the characteristics of the molecule. Secondly, Lp(a) concentration is more than 80% genetically determined, so that, unlike other cardiovascular risk factors, it cannot be regulated by lifestyle changes. Finally, although there are many promising clinical trials with specific drugs to reduce Lp(a), currently only PCSK9i (limited for use because of its cost) significantly reduces Lp(a).
However, and in line with other scientific societies, the SEA considers that, with the aim of increasing knowledge about the contribution of Lp(a) to cardiovascular risk, it is relevant to produce a document containing the current status of the subject, recommendations for the control of global cardiovascular risk in people with elevated Lp(a) and recommendations on the therapeutic approach to patients with elevated Lp(a).
La irrupción de la lipoproteína (a) (Lp(a)) en el estudio de los factores de riesgo cardiovascular es quizás, junto con el descubrimiento y uso de los fármacos inhibidores de la proproteína convertasa subtilisina/kexina tipo 9, (iPCSK9), la mayor novedad en el campo desde hace décadas. La concentración de Lp(a) (especialmente los niveles muy elevados) tiene una innegable asociación con determinadas complicaciones cardiovasculares, como los derivados de enfermedad vascular aterosclerótica (EVA) y o la estenosis aórtica. Sin embargo, existen varias limitaciones actuales tanto para establecer asociaciones epidemiológicas como para realizar un tratamiento farmacológico específico. En primer lugar, la medición de la Lp(a) depende en gran medida del test utilizado, principalmente por las características de la molécula. En segundo lugar, la concentración de Lp(a) está determinada en más del 80% por la genética, por lo que, al contrario de otros factores de riesgo cardiovascular no puede ser regulada con cambios del estilo de vida. Finalmente, aunque existen múltiples ensayos clínicos prometedores con fármacos específicos para reducir la Lp(a), actualmente solo los iPCSK9 (limitados para su uso por su coste) reducen de forma significativa la Lp(a).
Sin embargo, y en línea con otras sociedades científicas, la SEA considera que, con el objetivo de aumentar el conocimiento sobre la contribución de la Lp(a) al riesgo cardiovascular, se considera relevante la realización de un documento donde se recoja el estado actual del tema, las recomendaciones de control del riesgo cardiovascular global en las personas con Lp(a) elevada y recomendaciones sobre la aproximación terapéutica a los pacientes con Lp(a) elevada.
Kare Berg was the first to detect an antigen in serum strongly influenced by heredity, and found in low density lipoproteins (LDL) in patients with myocardial infarction, which he named apolipoprotein (apo)(a).1–3 Several studies confirmed that apo(a) is part of a lipoprotein (Lp) that otherwise resembles LDL. It is interesting to note that Lp(a) has only been identified in hedgehogs and primates, including humans.4
Structure, synthesis, and catabolismThe protein structure of Lp(a) essentially contains apolipoprotein B (apoB)-100 and apo(a), and it is precisely this combination that is the distinguishing feature of this lipoprotein.5 Cloning and sequencing of the apo(a) gene (LPA) in 1987 showed it to be a hydrophilic, highly glycosylated protein very similar to the plasminogen gene.6 Thus, it contains characteristic domains called kringles, which are structures of 80–90 amino acids organised in a triple loop. Apo(a) contains two types of kringle (K), IV and V, as well as a mutated protease domain lacking proteolytic activity.6 While there is only one copy of KV and of the protease domain in each apo(a) molecule, there are 10 KIV kringle subtypes, numbered 1–10 (KIV1–10). All of the domains, except KIV-2, have a single copy per molecule. KIV-2, however, varies in number of copies from one to more than forty, and is therefore a source of very highly polymorphic variation in the apo(a) of different individuals.
The number of KIV-2 kringles determines the total length and molecular weight of apo(a), which therefore varies between 300 and 800 kDa. Not only does the length and molecular weight of apo(a) depend on the number of copies of KIV-2, but KIV-2 is also an important inverse determinant of plasma Lp(a) concentration.7 Thus, the higher the copy number of KIV-2 in apo(a), the lower the plasma Lp(a) concentration in molar units.
The mechanisms of Lp(a) synthesis and catabolism are not yet fully understood. Lp(a) is considered to be predominantly synthesised by the liver. Interaction between lysine-rich domains between apoB-100 and apo(a) precedes the formation of a single disulphide bridge between the two apolipoproteins, which may occur extracellularly.8 Lp(a) also appears to be predominantly catabolised by the liver, although the receptors involved have not been clarified. The LDL receptor does not appear to play an essential role in the removal from the circulation of Lp(a).4 Some studies have identified small amounts of non-apoB-bound apo(a) fragments in urine, although the role of the kidney in Lp(a) catabolism not been well defined either.9
Genetic and environmental determinants of Lp(a) concentrationGenetic: number of KIV repeats and SNPLp(a) concentration is about 80% determined by codominant inheritance in the gene coding for Lp(a) (LPA).4,10 It has been estimated that 19%–70% of the variation in Lp(a) concentration depends on the number of KIV-2 repeats. Thus, short apo(a) isoforms (10–22 copies of KIV-2) are associated with 4–5 times higher Lp(a) concentration than long isoforms (>22 copies of KIV-2).4,11 This is due to a decrease in the efficiency of apo(a) synthesis in the long isoforms rather than to differences in catabolism. However, there can be more than 200-fold differences in serum Lp(a) concentration between unrelated individuals with similar length isoforms, and more than 2-fold differences in relatives with the same apo(a) isoforms).11 Thus, genetic variants other than the number of KIV-2 copies also determine Lp(a) concentration.12 Therefore, numerous single nucleotide polymorphisms (SNPs) that increase or decrease Lp(a) concentration have been described, such as the rs10455872 and rs3798220 SNPs.12 Interestingly, some of the SNPs located in the LPA gene are inherited in linkage disequilibrium with the number of copies of KIV-2.
It is also worth considering that up to 70% of the coding sequence is in the hypervariable region of KIV-2 not easily accessed by conventional sequencing technologies,13 and therefore this topic will require further study. Genetic variants at the APOE, CETP, and APOH loci may also, to a lesser extent, modulate plasma Lp(a) concentration. For example, the APOE2 allele is associated with lower Lp(a) levels.4,11
Ethnicity is also a determinant of Lp(a) concentration in serum, which is higher, in increasing order, in people of Chinese, Caucasian, South Asian, and Black ethnicity. These differences appear to depend primarily on the number of KIV-2 repeats.4,11 Lp(a) concentration is fairly stable throughout adulthood and slightly higher (5%–10%) in women than in men.4,11
Environmental: physiological (fasting, diet, exercise) and pathological (liver, kidney impairment, inflammation, and hormonal changes)Although not as well characterised as genetic factors, and generally of lesser importance, environmental factors can also modify serum Lp(a) concentration, especially in some physiological and pathological circumstances. There are also factors associated with Lp(a) concentration whose origin and clinical significance have not yet been fully identified. As an example, an inverse relationship between Lp(a) concentration and triglycerides has been identified, especially when triglycerides are higher than 400 mg/dL.14
Lp(a) concentration does not change substantially with fasting/postprandial states, or with exercise.11 Neither does diet seem to markedly influence Lp(a) concentration, with decreases of 10%–15% reported with low-carbohydrate and high-fat diets. Pregnancy doubles Lp(a) concentrations, while there is a slight increase in Lp(a) at menopause.11
As an organ of Lp(a) synthesis, severe liver impairment can alter Lp(a) concentration.11 Severe renal disorders tend to increase Lp(a) through increased synthesis associated with proteinuria or decreased catabolism,15 whereas in severe clinical situations, where there is a life-threatening acute-phase reaction, there is a decrease in Lp(a) concentration.16,17
Some hormones that affect lipoprotein concentration also affect Lp(a) concentration.11 Thus, thyroid disorders influence Lp(a) concentration in the same way as LDL-cholesterol. That is, hyperthyroidism decreases Lp(a) concentration, while hypothyroidism increases it. Growth hormone replacement therapy increases Lp(a) concentration and hormone replacement therapy in menopause lowers levels.11
To conclude this section, it is important to remember that, as mentioned above, changes due to hormones and acute phase reaction are relatively modest compared to levels that are genetically defined.
Pathogenic factors (arteriosclerosis)Although the serum concentration of Lp(a) is lower than that of LDL, there are indications that the pathogenicity of the former may be greater.18 This could be explained firstly by the tendency for Lp(a) to accumulate in the arterial wall, favoured by the binding of the lysine-rich domain of apo(a) KIV-10 to extracellular matrix proteins,19 and secondly by the ability of Lp(a) both to initiate vascular inflammation and to facilitate the progression of atherosclerotic damage.20 Oxidised phospholipids, which are mostly transported in serum by Lp(a), have also been posited as a major mediator of the proinflammatory and pro-calcifying mechanisms induced by Lp(a), either in atherosclerosis lesion of the vascular wall or at the level of the heart valves.21
Lp(a) may also have prothrombotic effects by increasing coagulation or inhibiting fibrinolysis (e.g., through its competition with plasminogen). However, potent changes in Lp(a) concentration induced by treatments do not affect ex vivo measures of fibrinolysis.22 In this regard, there is no epidemiological evidence of an association between elevated Lp(a) levels and increased risk of venous thromboembolism, a different context from atherosclerotic plaques.23
Laboratory methodology for Lp(a) testing in SpainA major challenge for the coming years in the presentation of Lp(a) as a predictor of cardiovascular disease (CVD) risk and potential therapeutic target is to establish a standardised methodology to test for it in clinical practice. This would allow comparisons to be made between different populations, and to establish consistent benchmark levels and recommendations. As we will see below, this has not yet happened, and there are several testing methods, and even different non-interchangeable units of measurement (mg/dL, nmol/L) that make interpreting laboratory results and the possibility of therapeutic interventions somewhat difficult. Lp(a) testing does not require any special conditions, and it can be performed fasting or non-fasting, although it is recommended not to test during acute processes other than acute coronary syndrome.24
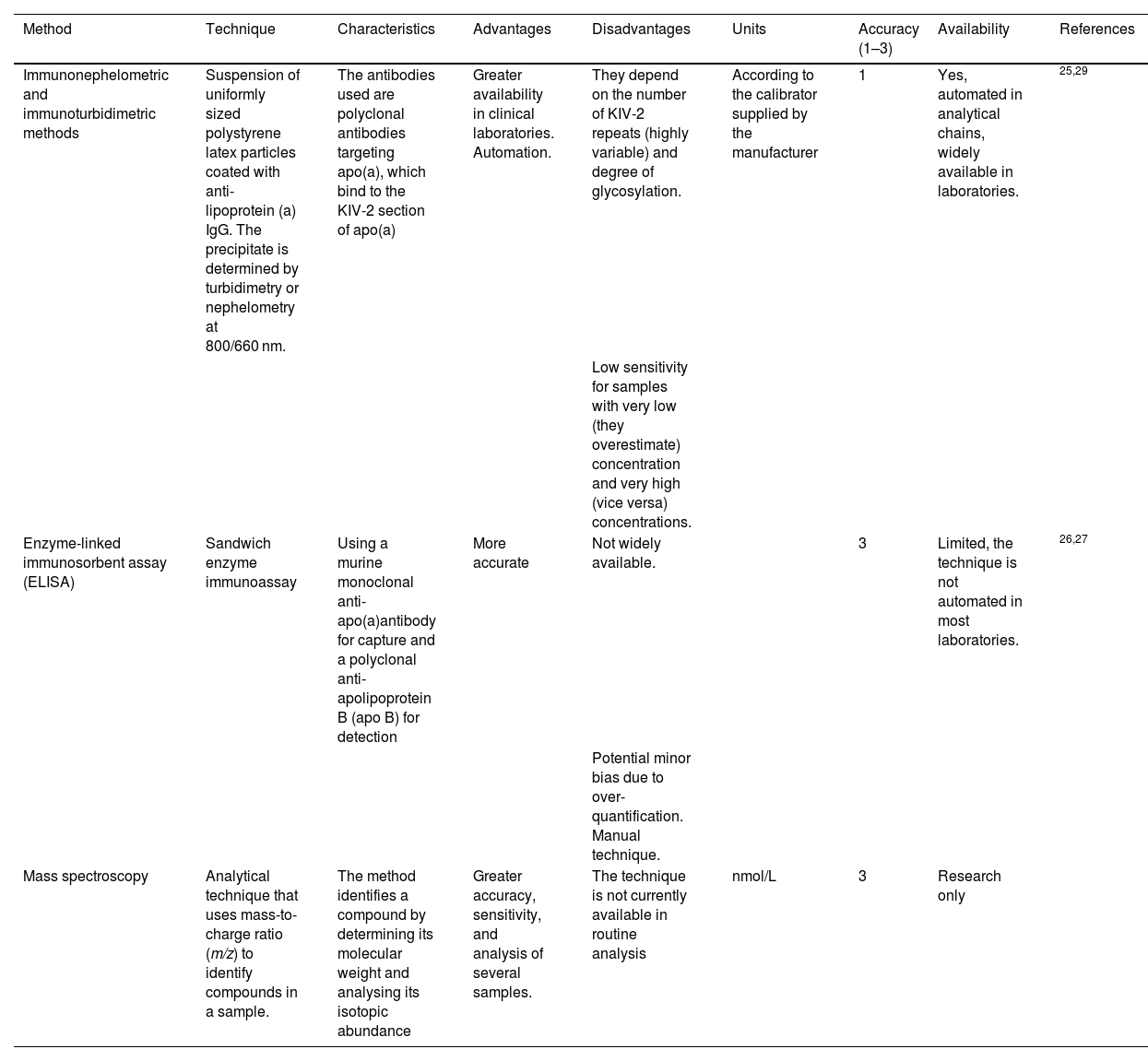
TechniqueTwo types of measurement methods are primarily used in Spain: those based on the detection of KIV-2 and those based on the measurement of ApoA and ApoB molecules (explained in greater depth in Table 1).
Tests available in Spain to determine Lp(a).
| Method | Technique | Characteristics | Advantages | Disadvantages | Units | Accuracy (1–3) | Availability | References |
|---|---|---|---|---|---|---|---|---|
| Immunonephelometric and immunoturbidimetric methods | Suspension of uniformly sized polystyrene latex particles coated with anti-lipoprotein (a) IgG. The precipitate is determined by turbidimetry or nephelometry at 800/660 nm. | The antibodies used are polyclonal antibodies targeting apo(a), which bind to the KIV-2 section of apo(a) | Greater availability in clinical laboratories. Automation. | They depend on the number of KIV-2 repeats (highly variable) and degree of glycosylation. | According to the calibrator supplied by the manufacturer | 1 | Yes, automated in analytical chains, widely available in laboratories. | 25,29 |
| Low sensitivity for samples with very low (they overestimate) concentration and very high (vice versa) concentrations. | ||||||||
| Enzyme-linked immunosorbent assay (ELISA) | Sandwich enzyme immunoassay | Using a murine monoclonal anti-apo(a)antibody for capture and a polyclonal anti-apolipoprotein B (apo B) for detection | More accurate | Not widely available. | 3 | Limited, the technique is not automated in most laboratories. | 26,27 | |
| Potential minor bias due to over-quantification. Manual technique. | ||||||||
| Mass spectroscopy | Analytical technique that uses mass-to-charge ratio (m/z) to identify compounds in a sample. | The method identifies a compound by determining its molecular weight and analysing its isotopic abundance | Greater accuracy, sensitivity, and analysis of several samples. | The technique is not currently available in routine analysis | nmol/L | 3 | Research only |
Immunoturbidimetric and immunonephelometric methods are the most widely used in our setting. The main characteristic of those available to most centres is that they bind to the KIV-2 section of the apo(a). As discussed above, KIV-2 is a sequence that may be represented in a higher or lower number of repeats within the apo(a) particle (see chapter 1), which may partially limit the accuracy of the test. These methods are evolving to be able to react with antibodies against a uniquely represented structure on the Lp(a) molecule, such as the KV or protease domain.25
Less available is the enzyme-linked immunosorbent assay (ELISA), which is theoretically more accurate. After trapping of Lp(a) with an antibody to apo(a), quantification is performed using an antibody to apo B, because there is a single molecule of each in the Lp(a) particle. This avoids inaccuracy in the measurement of variable sequences (which determine the different isoforms) within the apo(a) molecule,26 (Table 1). Some authors point out that assays of this type, based on apo(a) capture/apoB detection, may overestimate Lp(a) due to the marginal binding that Lp(a) particles may have noncovalently associated with triglyceride-rich lipoproteins.27
In relation to existing methods, and with reference to those based on KIV-2 detection, we should bear in mind that Lp(a) concentration is strongly genetically determined, and diet or lifestyle changes are of minimal influence. Part of the genetic influence on Lp(a) in offspring is the number of KIV-2 repeats (see section on Lp(a) genetics). Particles with a higher number of KIV-2 repeats are larger and heavier, an estimated mass difference of 19% has been reported between an Lp(a) particle with 6 KIV-2 repeats in apo(a) compared to Lp(a) with 35 KIV-2 repeats, and therefore the measurement of Lp(a) concentration may be partly artefactual when using assays that are not isoform independent.28 These measurement errors primarily affect quantifications with extreme results, underestimating the concentrations of samples from individuals with small Lp(a) isoforms (associated with higher Lp(a) levels) and vice versa.29 This is particularly important because thus the risk of patients with small Lp(a) isoforms and vice versa would be underestimated.11
Measurement units and conversionMost current consensus, therapeutic approaches, and clinical practice guidelines express Lp(a) measurements in mass units (mg/dL). This is mainly for didactic and historical reasons, as clinicians are more accustomed to using these units. Therefore, conversion systems from the original units expressed by the different analytical tests mentioned above are often used. However, in this case, this approach is not really metrologically correct. This is because these analytical tests only assess the concentration of the protein fraction of Lp(a), and not the mass of the other components of the Lp(a) particle, such as cholesterol, cholesterol esters, phospholipids, triglycerides, and carbohydrates. Furthermore, when converting nmol/L to mg/dL, a second metrological error is made, because the molecular weight of Lp(a) isoforms may vary, which does not allow one-to-one conversion between mass and molar units.
For practical purposes, in the rest of this document we will continue using the units commonly employed in the literature (mg/dL), although the reader should be aware that this approach produces some margin of error, especially in the extreme values of Lp(a). This is likely to improve with the introduction of more accurate, isoform-sensitive, molar apo(a)-based tests, with apo(a) test results reflecting the amount of Lp(a) particles present in an individual's serum/plasma, independent of the number of KIV-2 repeats, the degree of N- and O-glycosylation of apo(a) or the lipid composition of Lp(a). This will establish limits of normality and more precise cutoffs for different therapeutic actions to be expressed in the correct units (nmol/).29–33
Several ongoing initiatives in this direction are attempting to solve the metrological problems in more accurate measurement of Lp(a) by manufacturing calibrated materials in molar units traceable to verified references (WHO/IFCC, Northwest Lipid Metabolism, and Diabetes Research Laboratory (NLMDRL), International Federation of Clinical Chemistry and Laboratory Medicine (IFCC).34
To conclude, several papers have recently been presented supporting the standardisation of Lp(a) measurement using by mass spectrometry with the most advanced quality standards.35,36
Until these tests become a reality, the European Atherosclerosis Society (EAS) recommends that test-specific reference values be included in laboratory reports. Although, as noted above, conversion between units is not recommended, we recognise that sometimes clinicians need guidance on their patient's test result with respect to clinical practice guidelines and consensus papers. In this regard, conversion factors from mg/dL to nmol/L usually range from 1:2.0 to 1:2.5,11,37 although, as discussed, this conversion is not very accurate and should only be used for a rough approximation of the test result and probably only to determine whether a patient is in a very high or very low risk zone.38 Different conversion factors may therefore appear in different parts of this document and the reader should bear this in mine. This is because we have tried to respect the conversions made in the original articles.
In summary, and given the current heterogeneity of Lp(a) testing, we should stress some key concepts, for example, serum Lp(a) concentrations should ideally be measured using a method where the isoform size effect has been minimised using appropriate antibodies with certified calibrators that can trace Lp(a) values to the WHO/IFCC reference material, and if the practitioner's working laboratory does not have these methods, it should be understood that the accuracy of the test is lower. Results should also ideally be expressed in nmol/L of Lp(a) particles, and if other units are used, the reference limits of the test used should always be given. Finally, it is important to stress that conversion from mass units to molar units or vice versa introduces inaccuracy, and it should always be understood that the results obtained from it are only for guidance.
Lipoprotein(a) and its relationship to vascular disease, aortic stenosis, and other diseasesSince its discovery, many epidemiological studies have been published linking Lp(a) to the risk for atherosclerotic vascular disease (AVD).1 The first prospective studies, in the 1990s, did not find a consistent correlation with AVD or acute myocardial infarction (AMI), probably because inaccurate measurement methods were used.39 This resulted in a loss of scientific interest in Lp(a) in the following years, and publications on Lp(a) significantly decreased over that period. However, more recent epidemiological and genetic studies suggest a causal association between elevated Lp(a) levels, AVD, aortic stenosis (AS), along with other diseases. Most of the population has low or very low Lp(a) concentrations, but it is estimated that 20%–25% have levels higher than 50 mg/dL, which the European Atherosclerosis Society considers indicates an increased risk for CVD morbidity and mortality.11
Epidemiological studies linking Lp(a) to coronary heart diseaseLarge prospective general population studies were published in the first decade of this century. The Copenhagen City Heart Study followed 9339 Danish individuals for 10 years and found that rising Lp(a) concentrations were independently associated with the risk of coronary artery disease (CAD), increasing the risk of AMI by 9% for each 10 mg/dL increase in Lp(a) concentration40; thus, compared to an Lp(a) concentration < 5 mg/dL, an Lp(a) level ≥ 120 mg/dL would confer an almost 4-fold increased relative risk for infarction. The same study showed that a high Lp(a) level was associated with increased CV and all-cause mortality, regardless of low-density lipoprotein cholesterol (LDL-C) concentration.41 A 2009 meta-analysis of 36 prospective cohorts with 126,634 participants without prior AVD by the Emerging Risk Factors Collaboration confirmed a continuous and independent, albeit modest, association of Lp(a) concentration with risk of non-fatal AMI and CV death.42 Although this meta-analysis included mainly Caucasian studies, other racial groups were also represented, and no differences were found in the risk estimates for different ethnic groups. In the later Atherosclerosis Risk in Communities (ARIC) study in white and black subjects, Lp(a) levels were positively associated with AVD with equal strength in both racial groups, although median Lp(a) concentrations were 2–3 fold higher in the black subjects than in the white.43 Analysis of 460,506 middle-aged individuals from the UK Biobank database followed for a median of 11.2 years showed a linear association between Lp(a) concentration and risk for AVD (hazard ratio [HR] of 1.11 (95 % CI, 1.10–1.12) per 50 nmol/L increase.44
Also, in individuals with familial hypercholesterolaemia (FH), an increased risk of CAD has been observed in those with Lp(a) concentrations above 50 mg/dL when compared to individuals with FH and Lp(a) levels below 50 mg/dL.45
Elevated Lp(a) levels have also been associated with recurrence of CV events in individuals with established CAD, although the data in these populations have been inconsistent.46 In a Danish study in a secondary prevention setting, increasing Lp(a) concentration was associated with an increased risk of recurrence, and it was estimated that a 50 mg/dL (105 nmol/L) reduction over 5 years would be necessary to achieve a 20% reduction in the rate of CV events.47 In a meta-analysis of 18,976 subjects, Lp(a) levels > 80th percentile were predictive of recurrent CV events in CAD patients treated with statins and were associated with 40% more major CV events (MACE), but only when baseline LDL-C was ≥130 mg/dL.48
In short, epidemiological studies show that an increased Lp(a) concentration is associated with an increased risk of AVD, primarily CAD, being of greater magnitude in primary prevention populations. The association between genetic variants associated with elevated Lp(a) and CV events disappears when adjusted for Lp(a) levels, suggesting a direct causal role of Lp(a) in the development of CAD.49
Epidemiological studies linking Lp(a) to aortic stenosis (AS)Calcific AS is the most common form of acquired valvular heart disease requiring intervention. The prevalence of AS is estimated to be 2%–4% in individuals over 65 years of age and the number of people with an indication for aortic valve replacement is expected to double by 2050.50 In recent years, the understanding of the pathophysiological mechanisms behind the development of this disease has evolved from degenerative calcification to an active process resulting from the interaction of genetic factors and chronic inflammation, favoured by risk factors such as smoking, hypertension, and hypercholesterolaemia. Although risk factors between calcific AS and CAD overlap, ≈50% of patients with calcific AS do not have concomitant CAD. Furthermore, statins, which have a significant beneficial effect on CAD, have to date not been shown to halt the progression of AS,51,52 which suggests that other pathophysiological factors are involved in calcific AS.
A paper published in 2013 was the first to establish that Lp(a) levels derived from genetic variations in the LPA locus were associated with aortic valve calcification and clinical AS.53 A subsequent study in 77,680 participants, combining data from the Copenhagen City Heart Study and the Copenhagen General Population Study, showed that elevated Lp(a) levels were associated with an increased risk of AS, and that Lp(a) levels > 90 mg/dL predicted a threefold increased risk of AS.54 These findings have been corroborated in subsequent epidemiological studies. More recently, a systematic review of 21 studies, including case-control, prospective, and retrospective observational, and genetic studies, has shown a significant association between elevated Lp(a) and calcific AS; elevated Lp(a) also predicts accelerated haemodynamic progression of AS and an increased risk of aortic valve replacement, especially in young patients.55 Increased Lp(a) is associated with aortic valve calcification especially in younger individuals (45–54 years), in whom the risk triples when Lp(a) levels are above the 80th percentile versus lower levels (15.8% versus 4.3%).56 In a sub-study of the FOURIER trial, enrolling 27,564 AVD patients taking statin therapy, elevated Lp(a) levels, but not LDL-C levels, were associated with a higher risk of subsequent AS events, either progression or need for aortic surgery with valve replacement.57
Therefore, elevated Lp(a) concentration is associated with the development of AS, especially in young people, with a higher rate of progression and with an increased risk of valve replacement.
Epidemiological studies linking Lp(a) to heart failure (HF) and atrial fibrillationThe combined Copenhagen General Population Study and Copenhagen City Heart Study showed that Lp(a) levels above the 75th percentile and certain LPA risk genotypes (such as rs3798220 and rs10455872) increased the risk of AMI and AS, and that levels above the 90th percentile were associated with an increased risk of HF, which appeared to be partially mediated by the increased risk of AMI and AS.58
The association between AVD and atrial fibrillation is well known. However, it is not known whether Lp(a) is an independent causal factor for atrial fibrillation. In a study from the UK Biobank cohort, which recruited over 500,000 people aged 37–73 years across the UK between 2006 and 2010, the authors concluded that Lp(a) was implicated in the risk of developing atrial fibrillation, which was partly independent of the atherosclerotic effect.59
Epidemiological studies linking Lp(a) to atherosclerosis in other arterial territoriesThe relationship between Lp(a), stroke, and peripheral arterial disease (PAD) is pathophysiologically plausible, because atherothrombosis is the main aetiopathogenic basis of both pathologies, similar to coronary heart disease. Thus, the atherogenic effect of Lp(a) would be exerted in other arterial territories, either through the LDL that forms part of the Lp(a) molecule, the proinflammatory effect of the oxidised phospholipids it contains, or the antifibrinolytic and thrombogenic properties of apo(a).4
The relationship between stroke and CVD risk factors is more complex than for CAD or PAD. This is because stroke can have different aetiologies, including, in addition to atherothrombosis, haemorrhage, embolism, or arteriolar hyalinosis. Nevertheless, there is a large body of evidence from epidemiological studies showing that increased Lp(a) concentration is associated with an increased risk of ischaemic stroke. Large-scale studies show an association between Lp(a) concentration and stroke risk. In the Copenhagen General Population Study (n = 49,699) and the Copenhagen City Heart Study (n = 10,813)60 concentrations were gradually, directly, and continuously related to stroke risk. This relationship has been confirmed in large meta-analyses, including the Emerging Risk Factors Collaboration, with 1.3 million person-years of follow-up, in which a continuous association between Lp(a) concentration and stroke risk was observed.42 In this meta-analysis, a 3.5-fold higher Lp(a) concentration (per 1 standard deviation) was associated with a risk for ischaemic stroke of 1.10 (95% CI 1.02–1.18), which was independent of other CVD risk factors. Similarly, in a meta-analysis of 20 articles (n = 90,904 individuals), high Lp(a) concentrations were associated with an odds ratio (OR) of ischaemic stroke of 1.41 (95% CI 1.26–1.57) in case-control studies and 1.29 (95% CI 1.06–1.58) in prospective studies.61 Consistent with these data, Lp(a) concentrations are also associated with residual risk of stroke recurrence in patients after first ischaemic stroke,62 although the association may be attenuated in patients with low concentrations of LDL-C or inflammatory biomarkers.63
The association between Lp(a) and PAD has been demonstrated in prospective studies of large population groups, including the Edinburgh Artery Study,64 the Scottish Heart Health Extended Cohort Study,65 and the EPIC study.66 The latter was conducted in a large cohort of healthy individuals (n = 18,720) and a 2.7-fold increase in Lp(a) concentration was associated with an increased risk of this disease of 1.37 (95% CI 1.25–1.50), irrespective of LDL-C values.
In an analysis of the Spanish FRENA registry among 1503 stable outpatients with CAD, cerebrovascular disease, or PAD, those with Lp(a) concentrations between 30 and 50 mg/dL had a higher rate of limb amputations than those with concentrations below 30 mg/dL (Relative Risk [RR] 3.18; 95% CI, 1.36–7.44). The risk of amputation was even higher when the Lp(a) concentration was above 50 mg/dL (RR 22.7; 95% CI 9.38–54.9).67 Similarly, a systematic review including 493,650 subjects found that elevated Lp(a) was associated with an increased risk of intermittent claudication (RR 1.20), PAD progression (RR 1.41), restenosis (RR 6.10), death, and PAD-related hospitalisation (RR 1.37), limb amputation (RR 22.75), and lower limb revascularisation (RR between 1.29 and 2. 90 depending on factors such as Lp(a) cut-off points and years of follow-up), irrespective of traditional CVD risk factors.68
The higher the Lp(a) concentration, the higher the risk of clinical manifestations of atherosclerosis in more than one arterial territory, i.e., generalised atherosclerosis, and the greater the severity of the clinical manifestations of the disease,69 including higher all-cause mortality.70 Patients with acute coronary syndrome undergoing percutaneous coronary intervention who have PAD have higher Lp(a) concentrations than those without PAD.71 Similarly, patients undergoing PAD or carotid surgery who have an Lp(a) > 30 mg/dL have a threefold higher risk of stroke, AMI, or CVD-caused death (95% CI 1.5–6.3) than those with Lp(a) concentrations within the reference range.70 Increased Lp(a) is also associated with an increased risk of restenosis requiring reintervention or amputation after lower limb arterial revascularisation procedures.72 In these patients, elevated Lp(a) levels were associated with an increased risk of stroke, CAD- or CVD-related death. Finally, some case-control studies suggest that elevated Lp(a) levels are associated with aortic dissection.73
In conclusion, elevated Lp(a) is associated with an increased risk of atherothrombotic stroke and PAD, and an increased rate of progression and risk for amputation in patients with PAD.
Epidemiological studies linking Lp(a) to diabetesStudies conducted in different populations have suggested that there is an inverse association between Lp(a) concentrations and the risk of type 2 diabetes mellitus. However, this association is not linear over the entire distribution of Lp(a) values, but is strong at very low Lp(a) concentrations and no longer evident at medium to high concentrations.74 In a case-control study of 143,087 Icelanders, 10% of those with the lowest Lp(a) concentrations (<3.5 nmol/L) had a higher prevalence of type 2 diabetes mellitus (OR 1.44, P < .0001). However, in those with concentrations above the median, the risk of diabetes was not associated with Lp(a) concentrations.75 Another prospective study in the general population also found that Lp(a) concentrations in the lowest quintile (<10 nmol/L) were associated with an increased risk of diabetes and the risk further increased in those with even lower Lp(a) concentrations.76 Another 2017 meta-analysis,77 showed that subjects with low Lp(a) concentrations, between 3 and 5 mg/dL, had a 38% higher risk of diabetes than subjects with values between 27 and 55 mg/dL.
It appears that only part of the association of Lp(a) with diabetes is due to a causal relationship, and it has not been possible to define whether the relationship is due to Lp(a) concentrations, apo(a) isoform length, or a combination or interaction between the two. It is important to decipher whether there is a causal relationship between very low concentrations of Lp(a) and diabetes risk because, if there is strong evidence of a causal relationship, it would be worth considering whether lowering Lp(a) to very low concentrations with the new drugs under development could increase the risk of diabetes. However, data from epidemiological studies suggest that to increase the risk of diabetes, Lp(a) concentrations would have to be lowered to extremely low levels, which seems unlikely.74 However, it has been well demonstrated that excess Lp(a) predisposes to micro- and macrovascular complications in the diabetic population and analyses of large clinical trials with PCSK9i78 suggest that in the diabetic or pre-diabetic population the potential diabetogenic risk of lowering Lp(a) would be far offset by the CVD benefit of markedly decreasing atherogenic cholesterol.79
Genetic contribution of Lp(a) to the rate of CV complicationsAs we have seen above, 80%–90% of Lp(a) concentration is genetically determined. This makes it possible to relate certain genetic variants in the LPA gene, which are primarily responsible for the concentration of Lp(a) in plasma, to the rate of CV complications without this association being distorted by external factors, which would indicate a causal association.80 In addition to the isoforms associated with genetic variants that regulate the number of KIV-2 repeats, rs10455872 and rs3798220 are other variants that have been linked to increases in Lp(a).
Genetic contribution of Lp(a) and CADSeveral genetic association and Mendelian randomisation studies have shown an association between different LPA variants and the severity and extent of atherosclerosis,81 and the risk of developing vascular complications, primarily coronary heart disease. Many of these studies have been conducted in the Danish population, and the number of KIV-2 repeats has been found to be inversely associated with the risk of AMI.82 However, while the association with CV and all-cause mortality was observed with the number of repeats, this association was not observed with the rs1045587241 variant. In the same study, the amount of cholesterol in the Lp(a) particles was calculated. For a similar amount of cholesterol, the magnitude of the association of Lp(a) was more strongly associated with all-cause and CV mortality than that observed for LDL, implying that the mortality effect of high Lp(a) is greater than that explained by its cholesterol content.
Studies in other populations and large meta-analyses have corroborated these associations. In a meta-analysis of 40 studies, people with smaller apo(a) isoforms had double the risk of infarction and stroke versus those with larger isoforms, >22 KIV-2 repeats.83 In other studies evaluating genetic variants associated with increased Lp(a), genetically predicted Lp(a) elevation was shown to be associated with CAD.49,82 Genetic variants associated with high Lp(a) also predict CAD risk in subjects already treated with statins, regardless of the reduction in LDL-C that they induce.84
Other studies have shown that allele variants associated with small reductions in Lp(a) concentration protect against the development of CAD.85,86 In fact, it has been shown that for every 10 mg/dL difference in genetically determined Lp(a) concentration, the rate of CAD is reduced by 5.8%.
Genetic risk scores have been developed with several Lp(a) variants to improve prediction of the risk for CV complications.87 While their prediction is adequate, the strength of the association is significantly reduced when Lp(a) concentration is included in the equation, and therefore their utility may be limited, in addition to the particular difficulty of performing genetic studies of the hypervariable regions of KIV.12
In secondary prevention, genetic variants of LPA have not been shown to have an impact on mortality in CAD subjects.88
Genetic contribution of Lp(a) to aortic valve stenosisIn the EPIC study, the rs10455872 variant was associated with an increased risk of AS,89 which was corroborated in a meta-analysis that included data from the UK Biobank.90 In this paper, the risk of AS increased by 10%–30% for each 10 mg/dL increase in genetically determined Lp(a) concentration. In contrast, the rs3798220 variant, with a lower population frequency, has been inconsistently associated with risk for AS.90 In a large case-control study involving 3469 subjects with AS, both variants were associated with an increased risk of AS.91 The association of both polymorphisms has also been studied in a recent meta-analysis92 which showed that subjects with these genetic variants had faster progression and higher risk of clinical complications, including death.
Genetic contribution of Lp(a) to other vascular diseasesThe Multiancestry Genome-Wide Association Study of Stroke consortium (n ≤ 446,696), which analysed the relationship of 9 polymorphisms associated with Lp(a) concentrations with the risk of different stroke subtypes, associated variants with high Lp(a) concentrations with increased risk of large artery stroke.93 For each one standard deviation increase in Lp(a) concentrations, the OR of large artery stroke was 1.20 (95% CI 1.11–1.30, P < .001). However, the risk of small vessel stroke was lower (OR .92; 95% CI .88–.97; P = .001) and no association was observed for total ischaemic stroke or cardioembolic stroke. This study also found an inverse association between genetically determined Lp(a) concentrations and the incidence of Alzheimer's disease, an issue on which the data are controversial and may involve several confounding factors.93 The association between genetic variants and stroke risk has also been demonstrated in the Danish population, where the number of KIV repeats and the rs10455872 variant are associated with an increased stroke risk of 20% and 27%, respectively.60
Genetically elevated Lp(a) has also been associated with the risk of heart failure,58 an association partly mediated by the greater risk for CAD and AS.
The LPA gene has also been associated with the development of PAD in Mendelian randomisation and genome-wide association studies.23,94–96 In reference to the latter, the Million Veteran Programme analysed the association of 32 million variants of deoxyribonucleic acid (DNA) sequences with PAD (31,307 cases and 211,753 controls) in individuals of European, African, and Hispanic ancestry.95 The results were replicated in an independent sample of 5117 PAD cases and 389,291 controls from the UK Biobank. Nineteen PAD-associated loci were identified, including 18 previously unidentified loci. These included loci in the LPA gene, which supports the association of Lp(a) with lower limb atherosclerosis.
Therefore, Lp(a) should be considered a useful biomarker for assessing the risk of atherothrombotic stroke and PAD. With new drugs currently in development, Lp(a) is likely to be a therapeutic target in the primary and secondary prevention of these diseases.
Genetic contribution of Lp(a) to other diseasesA lower risk of diabetes97 has been reported with high concentrations of Lp(a), which seems to depend mainly on the genetic variant, in particular the number of KIV-2 repeats.
Finally, the association between Lp(a) and the risk of venous thromboembolic disease is controversial. While the number of KIV type 2 repeats has been inversely associated with the risk of venous thromboembolism,98 Mendelian randomisation studies do not seem to confirm this association.80
Measuring Lp(a): when and in whomThere are currently two reasons for testing Lp(a) levels: 1) to improve CV risk estimation in conjunction with other risk factors; 2) to assess an emerging risk factor, fairly stable over time, and identify the population, towards developing potential future therapeutic approaches. The absence of effective treatments to significantly reduce Lp(a) concentration (in particular, the absence of intervention studies with relevant clinical endpoints, specifically CV events) and the difficulty in standardising Lp(a) measurements has delayed the systematic inclusion of Lp(a) measurement in CVD prevention guidelines.11,99–104
Renewed interest in Lp(a) as an independent vascular risk factor and the expectations of pharmacologically reducing Lp(a) levels from early clinical trials have resulted in the progressive inclusion of Lp(a) testing in selected patients. Initial recommendations focused on specific circumstances: early CVD (personal or familial), especially in the absence of other CV risk factors, a family history of elevated Lp(a), familial hypercholesterolaemia, and poor lipid-lowering response to statins.11,99–104 Given the increased accessibility of Lp(a) measurement in conventional laboratories and the growing recognition of its prognostic value from observational studies, the current trend, initiated by the Canadian guidelines, is to include Lp(a) measurement as part of the first global CV risk assessment of all patients.11,102,105 Lp(a) testing is therefore included as part of the initial global CV assessment recommendations and should be assessed in the context of the patient's age, sex, presence of other CV risk factors, and family history of early CVD. Since Lp(a) levels are genetically conditioned and generally remain stable throughout life, a single measurement is usually considered sufficient for clinical decision-making.11,106 However, it is reasonable to consider "confirmatory" testing in some circumstances: very high levels, doubts about the reliability of the method used, coexistence with situations that may alter its concentration, such as advanced renal failure or nephrotic syndrome, or if the first test took place in childhood, pregnancy, significant hepatocellular/thyroid dysfunction, sepsis, or acute inflammatory process, as outlined in Table S1.
This is the approach accepted in the recent consensus on the basic lipid profile co-led by the Spanish Society of Arteriosclerosis (SEA), the Spanish Society of Laboratory Medicine, and the Spanish Society of Cardiology99 in collaboration with 15 scientific societies.
Testing of Lp(a) levels is necessary in patients in secondary prevention whose levels are not known, firstly, due to its prognostic value, which may suggest that preventive or therapeutic measures be reinforced, with PCSK9i for example.107 Testing may also serve to identify patients who are candidates for clinical trials or emerging treatments specifically aimed at reducing Lp(a) levels.
As mentioned above, Lp(a) levels are 80% genetically determined. It may be questioned whether Lp(a) genotyping has prognostic value over the direct measurement of circulating Lp(a). Although some genetic polymorphisms (rs10455872 and rs3798220 SNPs) are relatively frequent and associated with ‘small’ Lp(a) and higher circulating levels, more than 70% of the variability (>500 variants) is encoded by the hypervariable regions of KIV-2 repeats, and it is particularly difficult to test them with conventional sequencing technologies.11,12 Additionally other genes (APOE, CETP, APOH loci) are associated with changes in Lp(a) concentration. Finally, genotyping has not been shown to provide additional prognostic information to the measurement of Lp(a) levels and is therefore not considered necessary outside of research studies.108
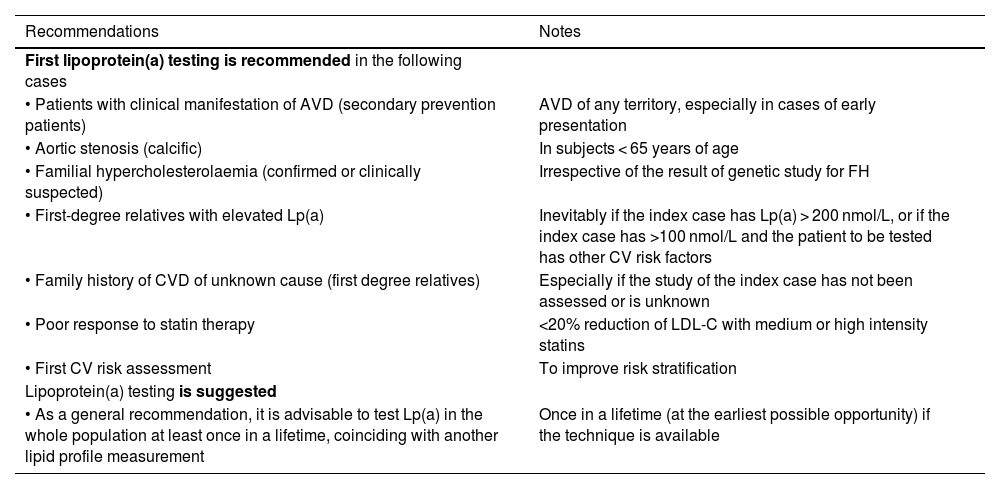
Table 2 summarises the SEA recommendations and suggestions for first Lp(a) testing.
Recommendations for first Lp(a) testing.
| Recommendations | Notes |
|---|---|
| First lipoprotein(a) testing is recommended in the following cases | |
| • Patients with clinical manifestation of AVD (secondary prevention patients) | AVD of any territory, especially in cases of early presentation |
| • Aortic stenosis (calcific) | In subjects < 65 years of age |
| • Familial hypercholesterolaemia (confirmed or clinically suspected) | Irrespective of the result of genetic study for FH |
| • First-degree relatives with elevated Lp(a) | Inevitably if the index case has Lp(a) > 200 nmol/L, or if the index case has >100 nmol/L and the patient to be tested has other CV risk factors |
| • Family history of CVD of unknown cause (first degree relatives) | Especially if the study of the index case has not been assessed or is unknown |
| • Poor response to statin therapy | <20% reduction of LDL-C with medium or high intensity statins |
| • First CV risk assessment | To improve risk stratification |
| Lipoprotein(a) testing is suggested | |
| • As a general recommendation, it is advisable to test Lp(a) in the whole population at least once in a lifetime, coinciding with another lipid profile measurement | Once in a lifetime (at the earliest possible opportunity) if the technique is available |
In general, given the stability of Lp(a) levels throughout life, it is not necessary to repeatedly test Lp(a) levels.11,106 However, it is reasonable to schedule a repeat test in clinical circumstances where there may be significant changes in Lp(a) levels (Table S1). The most marked changes may occur in advanced renal failure (particularly in peritoneal dialysis) and nephrotic syndrome,15,100 in which case retesting is more justified. In general, in all other clinical situations, changes are modest with a very modest impact on CV risk assessment, and therefore repeated measurement of Lp(a) is generally not considered necessary. For borderline values, a repeat test may be considered if the previous test was performed in childhood, pregnancy, hepatocellular dysfunction, in situations of frank hyper/hypothyroidism, sepsis, or major acute inflammatory processes and after menopause.11
Finally, it seems clear that repeat Lp(a) testing will be mandatory to assess response to treatment when available. Although the PCSK9i are currently the only lipid-lowering drugs in common use with a significant effect on Lp(a) (approximately 20% reduction),109 significant pharmacological development is underway with drugs specifically aimed at lowering its concentration, as will be outlined in the corresponding chapter of this document.
These recommendations and suggestions are summarised in Table S1.
The importance of including Lp(a) in CV risk assessmentAssessment of risk conferred by Lp(a): threshold versus continuous risk rateThe association between Lp(a) and CVD risk has been explored from two methodological standpoints. On the one hand, by trying to identify certain cut-off points or thresholds that distinguish risk categories, as has been done previously with other lipid particles, such as LDL-C, and, on the other hand, by assessing whether there is a continuous proportional relationship.
In the first approach, the risk conferred by elevated Lp(a) was assessed by defining different threshold or categorical concentrations. The Copenhagen City Heart Study82 found that from the 66th percentile of Lp(a) levels in 7524 subjects (equivalent to approximately 30 mg/dL), there is a significant increase in the risk of myocardial infarction over a 16-year follow-up.
In the second approach, the continuous relationship between Lp(a) and CV risk was analysed: one of the main studies in terms of sample size is from the UKBiobank44 with 460,506 middle-aged subjects (40–69 years) with a follow-up of 11.2 years. This study found that, in the total cohort, each 50 nmol/L increase in Lp(a) concentration led to an 11% increase in CVD risk (HR 1.11 for CAD and ischaemic stroke) in the Lp(a) range studied of up to 400 nmol/L. This figure varied slightly according to statin use, history of previous events and age, and was maintained for both sexes and different LDL-cholesterol concentrations.
Although there is sufficient evidence to argue that the relationship between Lp(a) levels and CVD risk is continuous, thresholds or cut-off points have been considered for practical reasons, similar to the CVD risk cut-off points that define the different risk categories based on LDL-C.
It is clear, however, that while the thresholds determine different “categories”, the physician should be aware of this continuous association of Lp(a) and CV risk, and adapt the therapeutic effort accordingly.
Risk calculators according to Lp(a)Of the many studies that have assessed the impact of elevated Lp(a) levels on CVD risk, the online calculator from the UK-Biobank-based study44 is of note, which estimates the future risk of ischaemic heart disease and stroke, up to the age of 80 years, based on the following variables: sex, age, total cholesterol, LDL-C, LDL-C, systolic blood pressure, treatment of arterial hypertension, body mass index, presence of diabetes, smoking, smoking cessation, and family history.110 Once the risk has been estimated, it can be adjusted to Lp(a) levels, for every 50 nmol/L increase, the risk is multiplied by 1.11. According to the authors of this website, the risk calculation is based on an artificial intelligence modality called Causal AI, generated through the participation of the company “DeepCausal AI” whose website currently has no content other than the logo (https://deepcausalai.org/) at the time of writing.
In a Danish cohort40 of 8720 general population participants recruited between 1991 and 1994 and followed up with no losses until 2011 assessing the incidence of AMI and CAD, a COX proportional hazards regression model was calculated for each type of event with traditional risk factors (age, sex, total cholesterol, HDL cholesterol, systolic blood pressure, smoking, and diabetes) and other models to which Lp(a) levels and/or Lp(a) risk genotype (KIV-2) and/or rs3798220 and rs10455872 polymorphisms have been added. The diagnostic performance of adding the new markers was assessed using the net reclassification index (NRI) and integrated discrimination index (IDI), and changes in the C-index. For 80th [Lp(a) ≥ 47 mg/dL] and 95th [Lp(a) ≥ 115 mg/dL] percentiles, the NRIs for myocardial infarction were 16% and 23%, and for CAD 3% and 6%, respectively. The authors provide beta coefficients for the regression models in their supplementary material, but these models have not been validated in other populations.
In the Italian cohort of the Bruneck study111 with 826 subjects from the general population whose risk was calculated according to Framingham and who had a Lp(a) concentration > 47 mg/dL added as an additional factor, a significant increased risk of CVD was observed (HR of 2.37). The addition of this factor therefore resulted in an improvement in NRI and IDI.
Improved risk classification of patients has also been examined by adding Lp(a) levels to the SCORE system. Using data from 16,777 subjects from the European Prospective Investigation of Cancer (EPIC), in whom risk was calculated with SCORE and modified to include the 30 mg/dL Lp(a) threshold (corresponding to the 80th percentile of that cohort), diagnostic discrimination was improved, especially in the intermediate risk group.112 In this group, the resulting NRI was 8.73%; this finding supports Lp(a) testing especially in intermediate-risk subjects in order to better adjust the therapeutic strategy.
None of the three previous studies provide complete risk assessment models, and therefore the only currently publicly available system for calculating CVD risk, including Lp(a), is online at www.Lp(a)clinicalguidance.com.
ThresholdsSeveral studies have assessed the CVD risk conferred by different Lp(a) thresholds. Thus Lp(a) values > 120 nmol/L in subjects without CVD confer a risk measured as a HR of 1.25 for PAD, 1.40 for CAD, 1.32 for AMI, 1.11 for ischaemic stroke, and 1.09 for CV mortality.87
Several consensus documents and clinical practice guidelines have recommended different risk thresholds in recent years. The National Lipid Association104 established that a threshold of 50 mg/dL or 100 nmol/L could be considered a factor that increases risk and therefore would recommend statin treatment. This value corresponds to the 80th percentile of the American Caucasian population. The American Heart Association and the American College of Cardiology consider thresholds of 50 mg/dL or 125 nmol/L as defining an increased risk of CVD.113 The UK consensus has established the following risk levels according to Lp(a) levels: low risk of CVD for levels between 30 and 90 nmol/L Lp(a), moderate risk between 90 and 200, high risk between 200 and 400, and very high risk for values > 400 nmol/L.100 The 2019 European guidelines already established that Lp(a) values > 180 mg/dL (430 nmol/L) confer a risk similar to that of patients with heterozygous familial hypercholesterolaemia.102 The European Atherosclerosis Society in its 2022 consensus document11 establishes a grey area between 30 and 50 mg/dL. Concentrations < 30 mg/dL (75 nmol/L) do not represent a significant increase in CVD risk, but >50 mg/dL (125 nmol/L) represent a significant increase. The grey area may be relevant in the presence of other risk factors. Although there is no direct conversion between mg/dL and nmol/L, some authors establish a multiplication factor between 2 and 2.5 in the conversion as mentioned in the corresponding section.
From these data we can see that there is an international consensus to define Lp(a) < 30 mg/dL (75 nmol/L) as a low-risk threshold and >50 mg/dL (125 nmol/L) as an increased risk threshold. The threshold of 180 mg/dL (430 nmol/L) would define a very high-risk equivalent to heterozygous FH.
Review of the impact of Lp(a) concentration on total CV riskThe epidemiology of CVD has taught us that serum Lp(a) concentrations do not follow a normal population distribution, showing a clear leftward shift. Therefore, and given that a large part of the general population has relatively low Lp(a) concentrations, clinical studies focus primarily on the upper tercile of Lp(a) concentration, where an increase of more than 20% in CV morbidity and mortality risk has been demonstrated.
It is clear from the above that Lp(a) is a risk factor for CVD in both primary and secondary prevention of CVD, even in the presence of low LDL-cholesterol levels. In the first clinical scenario, Lp(a) concentrations above the 75th percentile increase the risk of AS and AMI; levels > the 90th percentile are associated with an increased risk of heart failure,58 and only for extreme levels > the 95th percentile does the risk of ischaemic stroke and CV mortality increase.41,60,83 In the field of secondary prevention, three meta-analyses have linked elevated Lp(a) to an increased risk of severe CV events, although there is some inconsistency between studies.46,48,114 In patients with established CVD from the Copenhagen General Population Study 47 and AIM-HIGH,115 Lp(a) showed a significant association with the recurrence of severe coronary events. Furthermore, in a recent meta-analysis of individualised data from seven randomised, placebo-controlled trials of statin intervention in primary and secondary prevention, elevated baseline Lp(a) was independently and approximately linearly associated with CVD risk.116 Moreover, we should not forget that very high Lp(a) concentrations (>180 mg/dL [>430 nmol/L]) identify subjects with a lifetime CVD risk equivalent to untreated heterozygous FH.102
Data on the incorporation of Lp(a) levels in CVD risk estimatorsThe addition of Lp(a) to CVD risk prediction algorithms has shown variable effects, although the general conclusion is that, as with most biomarkers, it marginally improves risk discrimination. In a study of more than 28,000 women from the Women's Health Initiative, Women's Health Study and Justification for Use of Statins in Prevention (JUPITER), Lp(a) was associated with CVD only in those with baseline cholesterolaemia > 220 mg/dL, and improvement in prediction was minimal.117
Two studies have addressed reclassification of patients by Lp(a) quantification with prospective follow-ups of 15 and 6 years.111,118 The addition of Lp(a) reclassified 15%–40% of patients as high or low CV risk. More recently, Delabays et al.119 demonstrated in a low CV mortality country like Switzerland, that the addition of Lp(a) to the SCORE model refined CV prevention, particularly in intermediate risk individuals, with an 11.4% improvement in reclassification.
On this basis, the most recent EAS consensus statement11 proposes the Lp(a) concentrations that pose an increased risk for a given subject taking into account their overall CV risk. According to data obtained from the UK Biobank, an Lp(a) concentration of 50 mg/dL (115 nmol/L) increases the absolute CV risk of a person with an overall baseline CV risk of 10% over a lifetime by 4% and by up to 10% in a person with a baseline CV risk of 25%. However, an Lp(a) value of 100 mg/dL (230 nmol/L) would increase the absolute lifetime risk by 9.5 and 24% respectively.11 In other words, the impact of Lp(a) concentrations on CV risk is also conditioned by the individual's baseline global risk, and therefore a personalised clinical approach is required based on the characteristics of the individual concerned, and more intensive management of risk factors are recommended for the same Lp(a) concentration in patients who are already at high risk.
Finally, using the new Lp(a) risk calculator proposed by the consensus document of the European Atherosclerosis Society as outlined in section 5.2 of this chapter highlighted the following issues. Firstly, CVD risk will be considerably underestimated if Lp(a) concentration is elevated and not taken into account. Secondly, intervention on risk factors such as LDL-C cholesterol and blood pressure can mitigate at least part of global risk, even if Lp(a) levels are not changed. Moreover, since Lp(a) concentration is genetically determined, Lp(a) levels remain stable over time for most of the population. Therefore, in individuals with elevated Lp(a) concentrations, their lifetime burden will be very significant.
In conclusion, although there is no consensus on how to incorporate the risk attributable to elevated Lp(a) into conventional risk estimation models, testing Lp(a) levels may help to refine the estimation of CV risk at least semi-quantitatively, with a grey area (30−50 mg/dL), moderate (≈50−100 mg/dL), high (100−200 mg/dL), and very high (>200 mg/dL) increased risk.
Recommendations for re-estimation of CV risk with Lp(a)As previously mentioned, we do not have epidemiological studies in our setting that would allow us to estimate with confidence the additional risk of our patients with different Lp(a) levels. Add to this the lack of standardisation of Lp(a) testing methods and it is not possible to offer a strict quantitative approach. However, it is reasonable to attribute an increase in vascular risk similar to that estimated in the Caucasian population of the UK Biobank and use it as a corrective factor for the vascular risk estimated with the primary prevention tables (SCORE2, SCORE-OP).11,100 The corrective factor allows us to modify the estimation of risk and therefore set therapeutic targets for LDL-C control in accordance with the recalculated risk.
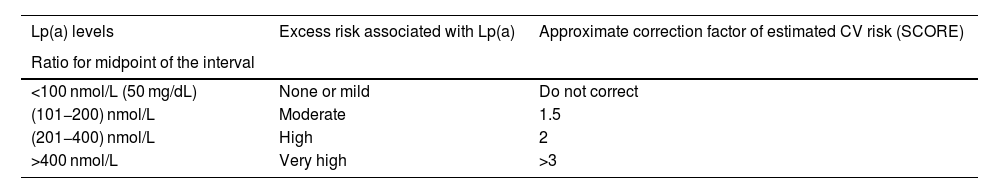
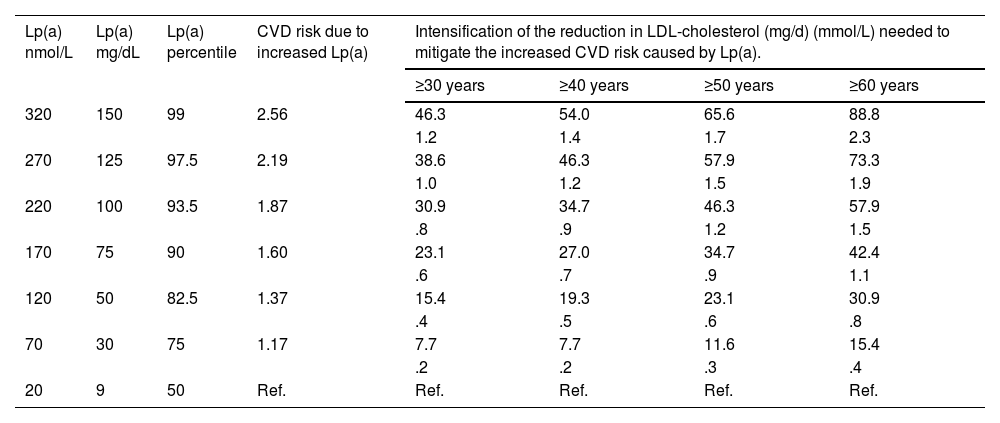
Based on the above information, Table 3 provides an approach to perform a semi-quantitative reassessment of CV risk by adding Lp(a), using correction coefficients calculated from the CV risk changes estimated in the UK Biobank for Caucasians with the British 'lifetime' risk scales. As indicated in the relevant section, a single conversion between molar units and mass of Lp(a) is not possible and therefore the correction coefficients should be used with caution. The Table is a semi-quantitative approach, and therefore the coefficients are an estimate of the mean values of the described ranges and can be adjusted somewhat more accurately as shown in Table S2.
Simplified model calculated from changes in CV risk estimated in the UK Biobank for Caucasians with the UK CV risk scales for 'lifetime' risk assessment. The proposed scale for Lp(a) is given in molar units and the mass units reported in the original consensus. As indicated in the relevant section, a single conversion between molar units and mass of Lp(a) is not possible and correction coefficients should be used with caution. The estimated CV risk to include excess risk due to Lp(a) is the risk estimation (e.g. SCORE2) multiplied by the correction coefficient of the Table. A more detailed model is provided in the supplementary material of this consensus.
| Lp(a) levels | Excess risk associated with Lp(a) | Approximate correction factor of estimated CV risk (SCORE) |
|---|---|---|
| Ratio for midpoint of the interval | ||
| <100 nmol/L (50 mg/dL) | None or mild | Do not correct |
| (101−200) nmol/L | Moderate | 1.5 |
| (201−400) nmol/L | High | 2 |
| >400 nmol/L | Very high | >3 |
In secondary prevention patients, elevated Lp(a) also confers an additional risk and can therefore be taken into account to identify patients at extreme CV risk in whom more ambitious targets for LDL-C control (<40 mg/dL) and the early use of particularly intensive treatments, such as PCSK9i, can be considered, as outlined in the SEA recommendations.107
As mentioned in the relevant section, there are currently no treatments available that have been shown to reduce cardiovascular complications through therapies to reduce Lp(a) levels. Therefore, clinicians and patients should be encouraged to participate in clinical trials to provide treatment based on the best scientific evidence in the coming years.
Finally, limited clinical evidence120 suggests that lipoprotein apheresis in CAD patients may improve their clinical outcome. There is no established indication for this therapeutic method, and it may be considered on an individual basis in patients with extremely high levels of Lp(a) with established CVD, or even lower levels in the case of progression of vascular lesions despite adequate control of other CV risk factors.
Finally, it should be borne in mind that regardless of the CV risk calculated without taking Lp(a) into account, several European guidelines have already expressed the increased risk conferred only by exposure to very high levels of Lp(a). Thus, patients with extreme Lp(a) levels, percentile > 99% (400 nmol/L; 200 mg/dL) should be considered per se at high baseline CV risk, even in the absence of other risk factors and therefore candidates for lowering LDL-C below 70 mg/d.100,102
Therapeutic targets and strategies to reduce Lp(a) levelsCurrent pharmaceutical approachThere is currently no approved pharmacological treatment to specifically reduce elevated plasma Lp(a) concentrations, and if the expected benefits based on observational evidence are confirmed in ongoing interventional studies, it will be a clinical necessity and a major challenge in the prevention and treatment of atherosclerosis. Current lipid-lowering drugs have a neutral or clinically insignificant effect on Lp(a) concentration (Table 4). However, in recent years, new therapies, some in the late stages of development, offer hope for the management of hyper-Lp(a).
Marketed drugs and their relationship with Lp(a) concentrations. Apolipoprotein B: apo B; Lipoprotein(a): Lp(a); Proprotein convertase subtilisin/kexin type 9: PCSK9.?: Uncertain.
| Drug group | Effect on Lp(a) | Mechanism of effect | Reduction in CVD |
|---|---|---|---|
| Resins | No | No | Yes (irrespective of Lp(a)) |
| Nicotinic acid | 20% reduction | Increase in hepatic apo-B catabolism | No |
| Statins | 8.5%–19.6% increase | Increase in hepatic LPA expression | Yes (irrespective of Lp(a)) |
| Lomitapide | 16% reduction | ? | ? |
| Fibrates | Variable and marginal | Reduction in triglycerides | No |
| PCSK9i | 25%–30% reduction | Unknown | Yes (dependent, in part, on Lp(a)) |
| Ezetimibe | No | No | Yes (irrespective of Lp(a)) |
| Bempedoic acid | 0%−5% increase | ? | Yes (irrespective of Lp(a))? |
| Obicetrapib | 35%–55% reduction | Increase in Lp(a) catabolism | ? |
Unlike other lipoprotein particles, such as LDL-C or triglyceride levels, plasma Lp(a) concentrations do not change in a clinically relevant way with changes in diet and healthy lifestyle.121 The effect of current lipid lowering agents on Lp(a) concentration is summarised in Table 4. While some of these drugs significantly reduce Lp(a) levels, there are no data on the potential CV benefit of this reduction.
Resins have not been shown to modify Lp(a) levels.122 Niacin produces a reduction in Lp(a) levels of around 20%.123,124 Previous studies have indicated that statin therapy can increase Lp(a) levels by a percentage. In this context, a recent meta-analysis, including 39 studies and 24,448 participants, shows that statin therapy did not change the CVD risk associated with slightly increased Lp(a) levels, differences in Lp(a) were not clinically significant.125 Given that the protective effect of LDL cholesterol-lowering therapy is proportional to baseline CV risk, the inclusion of Lp(a) in the risk estimation may identify patients with greater potential benefit because of their higher absolute risk. This suggests that patients at moderate or borderline CV risk and with elevated Lp(a) levels (>50 mg/dL) should have their risk recalculated and be considered for lipid-lowering therapy, initially with statins,104 and therapy in patients at high or very high risk should be intensified. In other words, elevated Lp(a) may suggest more ambitious LDL-C control targets. Lomitapide, whose current sole indication is homozygous FH, reduces Lp(a) levels in subjects with hypercholesterolaemia.126,127 The effect of fibrates is very variable and depends on the baseline triglyceride concentration.14 In a meta-analysis of seven RCTs of ezetimibe monotherapy, ezetimibe significantly reduced Lp(a) levels by 7.1%.128 However, another meta-analysis, ezetimibe, either in monotherapy versus placebo or in combination with statin versus statin alone, did not significantly affect Lp(a) levels.129 More recently, in a phase 2 trial, bempedoic acid did not significantly reduce Lp(a).130,131 Similarly, the combination of bempedoic acid with evolocumab was not superior to evolocumab monotherapy in terms of change in Lp(a) from baseline.132 In short, all these lipid-lowering drugs, either because of lack of efficacy, lack of significant clinical benefit, or because of the side effects of some of these molecules, are not indicated as tools to reduce high Lp(a) levels).133
The PCSK9i (evolocumab and alirocumab) have shown a marked effect on Lp(a) levels with a mean reduction of 26.7% (95% CI, −29.5% to −23.9%) with significant heterogeneity in relation to comparator and duration of treatment.134
Subgroup analyses of the Odyssey and Fourier studies show an absolute benefit of superior CV event reduction in patients with elevated Lp(a) after treatment with alirocumab and evolocumab, respectively. While some of the benefit may be attributable to the higher absolute risk in patients with elevated Lp(a), post hoc analyses suggest the possibility of an additional benefit linked to Lp(a) reduction per se, irrespective of the effect on LDL-C. The design of the studies and the post hoc nature of these analyses mean a definitive conclusion cannot be drawn in this regard.135,136
Inclisiran, a small interfering RNA (siRNA) targeting intracellular PCSK9, has been shown to reduce Lp(a) levels by approximately 20% (50%).137 The clinical benefit of this finding is currently unknown, although it could be hypothesised to be similar to that of PCSK9i. Finally, Obicetrapib, a selective cholesteryl ester transfer protein (CETP) inhibitor drug in clinical development, at doses of 5 mg and 10 mg has been shown to reduce Lp(a) by 33.8% and 56.5%, respectively.138
Failures in some clinical trials of drugs with presumed favourable effects on CVD due to improvements in the atherogenic lipid profile (including Lp(a) reduction) with niacin and CETP inhibitor drugs, call for caution before assuming CV benefits from Lp(a)-lowering drugs until adequate clinical trials have been developed that specifically assess relevant clinical endpoints, not simply 'improvements' in lipid profile.
ApheresisLipoprotein apheresis is the most effective intervention to date in decreasing Lp(a) concentrations.139 The efficacy of this intervention is remarkable, reducing Lp(a) levels by 70%–80%, but subsequently resulting in decreased mean interval concentrations of 25%–40%, depending on the course and baseline Lp(a) levels.139
A single-blinded controlled crossover trial in 20 patients with refractory angina and raised Lp(a) undergoing lipoprotein apheresis for 3 months suggests that this intervention may improve myocardial perfusion and improve angina control.140
An ongoing MultiSELECt RCT is comparing weekly Lp(a) apheresis with maximally tolerated lipid-lowering therapy in approximately 1000 patients aged 18–70 years with Lp(a) levels > 120 nmol/L or higher, LDL-C levels < 100 mg/dL, and CVD. The endpoint is to explore the clinical benefit of Lp(a) apheresis on MACE.120
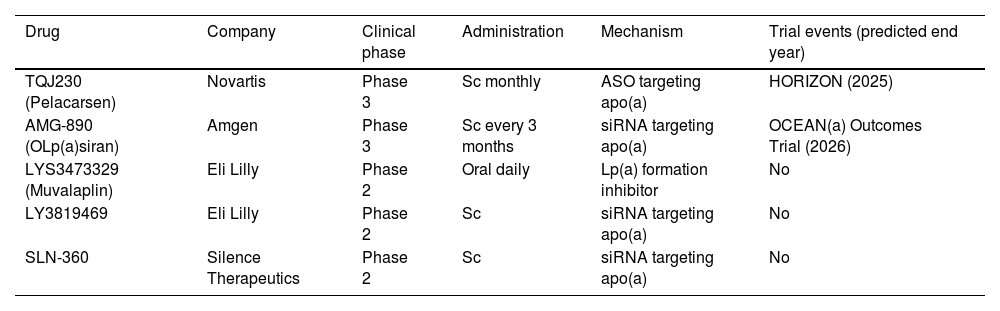
New therapies under developmentA completely different approach to reducing abundant proteins involves blocking their production by inhibiting the translation of their mRNA. The 2 main classes of RNA-targeting drugs developed for this purpose are single-stranded antisense oligonucleotides (ASOs) and double-stranded small interfering RNA (siRNA) (Table 5). Both strategies share a similar mechanism of action: after parenteral administration and introduction into the hepatocyte, they bind to a specific sequence of the mRNA of interest, at the nuclear level for ASOs and in the cytoplasm for siRNA. This interaction leads to degradation of the target mRNA and, therefore, decreased translation of the coded protein. One of the main advantages of RNA-based drugs is the possibility to target with high specificity proteins with high plasma concentrations such as apo(a), a component of the Lp(a) particle.141Pelacarsen is a 20-nucleotide synthetic ASO, conjugated to GalNAc3, and targeting the mRNA of apo(a), up to 80%, which has been shown in phase 1 and 2 trials.141,142 The impact of 80 mg monthly Pelacarsen on MACE reduction is being evaluated in the HORIZON trial (NCT04023552), a Phase 3 RCT involving more than 7000 patients with CVD and Lp(a) > 70 mg/dL, to compare its efficacy and safety. The results are expected in 2025.143OLp(a)siran, a siRNA that disrupts apo(a) production through degradation of apo(a) messenger RNA, inhibits Lp(a) assembly in the hepatocyte. In preclinical development, it has demonstrated a reduction in Lp(a) of up to 98% in patients with levels > 70 nmol/L without serious adverse events.144 In a phase 2 RCT, OCEAN(a) DOSE (OLp(a)siran trials of CV Events And lipoproteiN(a) reduction) 281 subjects with CVD and Lp(a) levels > 150 nmol/L assigned to receive OLp(a)siran (10 mg every 12 weeks, 75 mg every 12 weeks, 225 mg every 12 weeks, or 225 mg every 24 weeks) obtained significant reductions in Lp(a) levels (70.5%, 97.4%, 101.1%, and 100.5%, respectively).145 The Phase 3 RCT, OLp(a)siran trials of CV Events and lipoproteiN(a) reduction (OCEAN(a)) - Outcomes Trial (NCT05581303)146 is in the active recruitment phase. Another siRNA, SLN360 targeting apo(a) synthesis, has achieved reductions between 46% and 98% with therapy administered subcutaneously at a single dose of: 30 mg, 100 mg, 300 mg, or 600 mg in patients with Lp(a) > 150 nmol/L.147 There is also another ongoing Phase 2 RCT on SLN360 (Study to Investigate the Safety, Tolerability, PK and PD Response of SLN360 in Subjects with Elevated Lipoprotein(a); NCT04606602) pending release of results.148 In the ALP(A)CA study, the main purpose is to determine the efficacy and safety of LY3819469 in adults with elevated Lp(a.149 Finally, Eli Lilly has a daily oral small molecule inhibitor (LYS3473329 (Muvalaplin)), which inhibits Lp(a) formation, with promising results. Future approaches with oral therapies and even gene-editing treatments with nanoparticles capable of modulating Lp(a) transcription and translation are the first step towards definitive control of residual CV risk favoured by this lipoprotein.150 All of this is summarised in Table 5.
Drugs under study in the reduction of Lp(a). Apo(a): Apoprotein(a); ASO: Antisense oligonucleotides; LP(a): Lipoprotein(a); SC: Subcutaneous; siRNA: double-stranded small interfering RNA.
| Drug | Company | Clinical phase | Administration | Mechanism | Trial events (predicted end year) |
|---|---|---|---|---|---|
| TQJ230 (Pelacarsen) | Novartis | Phase 3 | Sc monthly | ASO targeting apo(a) | HORIZON (2025) |
| AMG-890 (OLp(a)siran) | Amgen | Phase 3 | Sc every 3 months | siRNA targeting apo(a) | OCEAN(a) Outcomes Trial (2026) |
| LYS3473329 (Muvalaplin) | Eli Lilly | Phase 2 | Oral daily | Lp(a) formation inhibitor | No |
| LY3819469 | Eli Lilly | Phase 2 | Sc | siRNA targeting apo(a) | No |
| SLN-360 | Silence Therapeutics | Phase 2 | Sc | siRNA targeting apo(a) | No |
Other clinical trials of interest currently under development are shown in Table S3.
Expected benefits in the reduction of Lp(a)We do not have any studies that have demonstrated the clinical benefit of Lp(a) reduction. This does not preclude us from estimating the expected benefit of Lp(a) reduction with drugs based on the risk attributable to Lp(a) in epidemiological studies and the reduction of Lp(a) with different interventions. In this regard, the exercise from Mendelian randomisation studies in parallel with the known effects of LDL lowering is interesting. Burgess et al. estimate that a reduction of approximately 100 mg/dL Lp(a) may be required to produce a benefit similar to that achieved by lowering LDL-C by 1 mmol/L (≈40 mg/dL).151 This exercise, however theoretical, offers interesting insights. Firstly, it gives us an estimate of how intensive LDL-lowering treatment might counteract the 'excess' risk attributable to increased Lp(a). We can see how an easily achievable reduction in LDL-C (e.g. 40% reduction in a patient with LDL = 100 mg/dL) would require a much more intense reduction (e.g. >60% reduction in Lp(a) in a patient with Lp(a) of 150 mg/dL) for an equivalent risk reduction. Therefore, Lp(a) reductions must be particularly intense to achieve significant clinical benefit. Modest reductions (e.g. 20% obtained with PCSK9i would therefore be of limited benefit. Fortunately, phase 2 studies with ASO and siRNA indicate that significant reductions (>75%) can be achieved with well-tolerated treatments. The risk/benefit balance in terms of CV events and safety must await the conclusion of correctly designed clinical trials. Therefore, patients with elevated Lp(a) and their physicians should be encouraged to participate in these trials so that appropriate treatment can be offered in the coming years.
The management of patients with high Lp(a): why increase control of other CVD risk factors in people with high Lp(a) levels?MotivationAs mentioned above, the association of elevated levels of plasma Lp(a) and increased risk of both AVD and aortic valve damage has been well established. The EAS recommendations on Lp(a) published in 2010,152 based on data from the Copenhagen General Population Study,82 established a single threshold level of risk, 50 mg/dL (115 nmol/L), above which it was considered necessary to reconsider the patients’ global risk. The SEA 2024 standards for the global management of CV risk153 also include this threshold to indicate the additional risk conferred by high Lp(a) concentrations. Current data suggest that the increased risk associated with Lp(a) concentrations is direct and continuous.154
Prevalence of elevated Lp(a) levels in SpainThere is currently little evidence on the prevalence of elevated Lp(a) in Spain. In a study conducted in the Canary Islands in 2012, screening of 1030 people found a prevalence of values above 47 mg/dL of 13.7%.155 There are other studies that have evaluated the prevalence of elevated Lp(a) in the results of analyses requested in clinical practice. In general, the prevalence of elevated Lp(a) (>50 mg/dL) in these studies is higher (25%–30%), although the data generally come from populations selected due to high CV risk or suspected high Lp(a) levels (relatives of people with elevated Lp(a), people in secondary prevention or at high CV risk, etc.) and therefore may be overestimated due to selection bias.156–158
CV risk reduction according to actual riskThe latest European consensus on Lp(a) suggests how much the additional reduction in LDL-C should be to reduce global CVD risk due to increased Lp(a) concentration, based on UK Biobank data11 (Table 6). Because this calculation was established on the basis of CV risk over a lifetime, the age factor is also very important, as it represents different periods of exposure to elevated Lp(a) concentrations. Thus, in a patient with an Lp(a) of 100 mg/dL (about 220 nmol/L), to nullify the CV risk attributed to that Lp(a) concentration, LDL-C would have to be reduced by about 31 mg/dL (.8 nmol/L) if they were 30 years old, or by about 58 mg/dL (1.5 nmol/L) if they were 60 years old (Table 6).11 Therefore, this Table gives practical information to estimate how much to intensify lipid-lowering treatment according to our patients’ Lp(a) levels. As discussed above (section 5.6), an extreme Lp(a) (>99% percentile; 400 nm/L; >200 mg/dL) has been identified in several European guidelines as equivalent to FH in target LDL, and therefore LDL should be reduced to <70 mg/dL.100,102
Increase in CVD risk according to Lp(a) concentrations and degree of LDL-cholesterol reduction to mitigate this increase, depending on the patient’s age. Taken from11 modified. We have added a column with the approximate conversion of Lp(a) concentration from nmol/L to mg/dL and conversion of LDL-cholesterol from mmol/L to mg/dL.
| Lp(a) nmol/L | Lp(a) mg/dL | Lp(a) percentile | CVD risk due to increased Lp(a) | Intensification of the reduction in LDL-cholesterol (mg/d) (mmol/L) needed to mitigate the increased CVD risk caused by Lp(a). | |||
|---|---|---|---|---|---|---|---|
| ≥30 years | ≥40 years | ≥50 years | ≥60 years | ||||
| 320 | 150 | 99 | 2.56 | 46.3 | 54.0 | 65.6 | 88.8 |
| 1.2 | 1.4 | 1.7 | 2.3 | ||||
| 270 | 125 | 97.5 | 2.19 | 38.6 | 46.3 | 57.9 | 73.3 |
| 1.0 | 1.2 | 1.5 | 1.9 | ||||
| 220 | 100 | 93.5 | 1.87 | 30.9 | 34.7 | 46.3 | 57.9 |
| .8 | .9 | 1.2 | 1.5 | ||||
| 170 | 75 | 90 | 1.60 | 23.1 | 27.0 | 34.7 | 42.4 |
| .6 | .7 | .9 | 1.1 | ||||
| 120 | 50 | 82.5 | 1.37 | 15.4 | 19.3 | 23.1 | 30.9 |
| .4 | .5 | .6 | .8 | ||||
| 70 | 30 | 75 | 1.17 | 7.7 | 7.7 | 11.6 | 15.4 |
| .2 | .2 | .3 | .4 | ||||
| 20 | 9 | 50 | Ref. | Ref. | Ref. | Ref. | Ref. |
The EPIC-Norfolk study assessed how people with elevated Lp(a) could reduce their CV risk by controlling other risk factors.159 The 14,000 participants were stratified according to scores on seven CV health parameters (body mass index, healthy diet, physical activity, smoking, blood pressure, diabetes, and cholesterol concentration) into three groups: healthy, intermediate, and unhealthy. After 11.5 years of follow-up, among the patients with Lp(a) concentrations ≥ 50 mg/dL, those in the healthy CV health score group had 67% fewer CV events than those in the unhealthy group. Although Lp(a) concentration was little influenced by CV risk factors, good CV health could substantially reduce the risk associated with elevated Lp(a) levels. Therefore, the lack of a proven treatment to specifically reduce Lp(a) and CVD should not be an obstacle (quite the contrary) to intensifying lipid-lowering therapy, adopting healthy lifestyles, maintaining ideal weight, avoiding smoking, controlling blood pressure and blood glucose.
Statins should be used primarily as lipid-lowering therapy. While some authors have reported a slight increase in Lp(a) with the use of statins, a recent meta-analysis of 39 studies and more than 20,000 patients found no clinically significant difference in Lp(a) concentration compared to placebo in patients at risk of CVD. However, PCSK9i in addition to reducing LDL-C by 60%, decrease Lp(a) levels by about 25%, and patients with higher baseline Lp(a) levels experience greater absolute reductions in Lp(a) and tend to achieve the greatest coronary benefit from PCSK9 inhibition.136,160
As mentioned above, there is a website (http://www.Lp(a)clinicalguidance.com) where lifetime CVD risk can be calculated, including classical risk factors and Lp(a) concentration. On the same website, it is possible to calculate how much this risk can be improved for specific decreases in LDL-C and/or systolic blood pressure.
Finally, in addition to intervening on risk factors in subjects with elevated Lp(a), it may be of interest to reduce their thrombotic risk with antiplatelet therapy. The ASPREE study evaluated the usefulness of 100 mg/day aspirin versus placebo in 19,000 subjects over 70 years of age in primary prevention with negative results after 5 years of follow-up. In almost 13,000 of these subjects, the rs3798220 polymorphism was identified, the C allele of which was associated with elevated Lp(a) concentrations and an increased incidence of CVD in the placebo group. When comparing the incidence of CVD in carriers of this allele, those who received aspirin had a significant decrease in CVD compared to the placebo group.161 Therefore, pending clinical trials to confirm these data, the results of this observational study would suggest that primary prevention subjects with elevated Lp(a) may benefit from preventive aspirin therapy.
Conclusions- •
This SEA consensus is made in view of the increasingly strong evidence associating Lp(a) with CV risk, especially at the 100 and 200 nmol/L, or 50 and 100 mg/dL thresholds.
- •
It is essential to move towards standardised Lp(a) testing, both in methodology and units of measurement.
- •
Lp(a) generally tends to remain at a similar concentration over time and most of it is genetically conditioned, and therefore lifestyle interventions have little impact on its levels.
- •
Given its stability over time, the SEA suggests universal Lp(a) testing once in a lifetime to coincide with the first biochemical measurement including lipid profile.
- •
The SEA recommends immediate Lp(a) testing, if it has never been performed before, in the presence of CVD, aortic stenosis, familial hypercholesterolaemia, first-degree relatives with elevated Lp(a), or early CVD of uncertain cause, or in patients with poor lipid-lowering response to statin therapy.
- •
Repeat Lp(a) testing is recommended in specific cases (see recommendations in the corresponding section)
- •
In the absence of specific clinical trials on Lp(a) reduction and cardiovascular events, all the current evidence indicates that Lp(a) may be the most important player to emerge in the field of lipidology in the last 50 years.
- •
The SEA recommends tailoring therapeutic effort to Lp(a) concentration in all patients.
- •
The emergence of risk calculators that include Lp(a), such as the calculator at www.Lp(a)clinicalguidance.com, can help the healthcare professional in estimating global CVD risk.
- •
The same document provides tables for CV risk reassessment adapted to Lp(a) concentration.
- •
The PCSK9i are the only drug group that has to date demonstrated some therapeutic power to reduce Lp(a.
- •
This document also contains a table to consult on intensifying LDL-C therapy to counteract the effect of elevated Lp(a).
- •
The results of clinical studies on lowering Lp(a) levels in CVD prevention may change the position of the SEA.
Some authors have received honoraria from different pharmaceutical companies for their participation in conferences and consultancies, which are detailed in the corresponding section. The authors have not received any remuneration for this report and have no other conflict of interest to declare.
Dr Delgado-Lista has received honoraria for conferences, educational events and meeting support from Amgen, Novartis, Ferrer, Laboratorios Dr Esteve, Menarini, Novartis, Novo-Nordisk, Sanofi, MSD, Servier, AstraZeneca, Mylan.
Dr Mostaza JM has received honoraria for consultancy work, as an expert witness, for lectures, Advisory Boards, presentations, manuscript, and educational events from Almirall, Alter, Amgen, Daiichi-Sankyo, Ferrer, Novartis, Sanofi, Servier, Viatris, and Ultragenix.
Dr Arrobas-Velilla T has received honoraria for consultancy from Amgen; Sanofi; Amarin; Daiichi; Novartis, for lectures, presentations, manuscript, and educational events and meetings from Amgen; Sanofi; Mylan; Sevier; Amarin; Amryt; Daiichi; Novartis.
Dr Blanco Vaca J has received lecture fees from Daiichi-Sankyo and European Atherosclerosis Society, and financial support for course organisation from Amarin, Daiichi-Sankyo, MSD, Novartis, and Sanofi.
Dr Masana L has received honoraria for lectures or consultancy work from Amarin, Amryt, Daiichi-Sankyo, Novartis, MSD, Sanofi, Servier, Ferrer, and Ultragenix.
Dr Pedro-Botet J has received for consultancy work, expert testimony, lectures, Advisory Boards, presentations, manuscript or educational events, meeting support from Amarin, Amgen, Daiichi-Sankyo, Esteve, Ferrer, MSD, Novartis, Sanofi, and Viatris.
Dr Perez-Martinez P has received honoraria for lectures, presentations, and educational events and meetings from Ferrer, Novo-Nordisk, Amgen, Daiichi Sankyo, Esteve, and Viatris.
Dr Civeira F has received honoraria for conferences and Advisory Boards from Amgen, Sanofi, Novartis, and Ferrer.
Dr. Cuende Melero JI has received honoraria for consultancy work, as an expert witness, for lectures, Advisory Boards, presentations, manuscript, educational events, meeting support, and contracts from MSD, Daiichi-Sankyo, Sanofi, Bayer, and Almirall.
Dr. Gómez Barrado JJ has received honoraria for consultancy work, expert testimony, lectures, Advisory Boards, presentations, manuscript or educational events, meeting support from Amarin, Amgen, Novartis, Ferrer, Daiichi-Sankyo, Sanofi, Bayer, and Almirall.
Dr Lahoz C has received honoraria for consultancy work, as an expert witness, for lectures, Advisory Boards, presentations, manuscript and educational events, meeting support from Amarin, Alter, Novartis, Daiichi-Sankyo, and Sanofi.
Dr. Pintó X. has received honoraria for consultancy work, as an expert witness, for lectures, Advisory Boards, presentations, manuscript and educational events, meeting support from Akcea, Almirall, Amarin, Amgen, Daiichi-Sankyo, Ferrer, Menarini, Novartis, Organon, Sanofi, Sobi and Viatris.
Dr Suarez Tembra M has received lecture fees from Laboratorios Servier and Daichi and support for congress attendance from Amgen, Servier, Daichi and Alter.
Dr Lopez-Miranda J has received honoraria for consultancy work from Amgen and Sanofi, and for conferences, presentations, manuscript and educational events, and meetings from Amgen, Sanofi, Novartis, Laboratorios Dr Esteve: Laboratorios Ferrer.
Dr. Guijarro C: has received honoraria for consultancy work, for conferences, Advisory Boards, presentations, manuscript, and educational events, from Amarin, Amgen, Daiichi-Sankyo, Ferrer, Novartis, MSD, Sanofi, and Servier.
The following is Supplementary data to this article: