




La superfamilia de anexinas está constituida por 12 proteínas con alta homología estructural que se unen a fosfolípidos de membrana de una manera dependiente de Ca2+. Diferentes estudios de sobreexpresión, inhibición o usando proteínas recombinantes han identificado que la función principal de estas proteínas está relacionada con su unión dinámica y reversible a membranas. Estas proteínas se encuentran en múltiples compartimentos celulares participando y regulando diferentes funciones como el tráfico de membranas, el anclaje al citoesqueleto celular, la regulación de canales iónicos, así como actividad proinflamatoria o antiinflamatoria y anticoagulante. El uso de animales deficientes en alguna de estas anexinas ha permitido establecer sus posibles funciones in vivo, demostrando que las anexinas pueden participar en funciones relevantes independientes de la señalización por Ca2+. En esta revisión nos centramos principalmente en el papel que juegan las diferentes anexinas en el remodelado vascular patológico que subyace a la formación de la lesión aterosclerótica así como en el control de la homeostasis del colesterol.
The annexin superfamily consists of 12 proteins with a highly structural homology that binds to phospholipids depending on the availability of Ca2+-dependent. Different studies of overexpression, inhibition, or using recombinant proteins have linked the main function of these proteins to their dynamic and reversible binding to membranes. Annexins are found in multiple cellular compartments, regulating different functions, such as membrane trafficking, anchoring to the cell cytoskeleton, ion channel regulation, as well as pro- or anti-inflammatory and anticoagulant activities. The use of animals deficient in any of these annexins has established their possible functions in vivo, demonstrating that annexins can participate in relevant functions independent of Ca2+ signalling. This review will focus mainly on the role of different annexins in the pathological vascular remodelling that underlies the formation of the atherosclerotic lesion, as well as in the control of cholesterol homeostasis.
Las enfermedades cardiovasculares (ECV) son la principal causa de mortalidad en los países desarrollados. Aunque la tasa de mortalidad debida a causas cardiovasculares ha disminuido considerablemente en las últimas dos décadas, 17,3 millones de personas fallecen anualmente por ECV y se estima un crecimiento hasta los 23,6 millones de personas para el año 20301. El término ECV se usa para describir diferentes patologías que afectan al corazón y al sistema circulatorio, incluyendo fallo cardiaco, enfermedad de las arterias coronarias, ictus, hipertensión y aterosclerosis, entre otras. En particular, la aterosclerosis se refiere al desarrollo de placas ateromatosas en el revestimiento interno de las arterias. Las células endoteliales del vaso, que generalmente son resistentes a la unión de leucocitos, expresan moléculas de adhesión que permiten el anclaje de los glóbulos blancos en la superficie celular cuando están en presencia de estímulos que las activan, como pueden ser la dislipemia, la hipertensión o las moléculas proinflamatorias. Los cambios que ocurren en paralelo en la permeabilidad endotelial y en la composición de la matriz extracelular promueven la entrada y la retención de lípidos, en particular lipoproteínas de baja densidad (LDL), en la pared arterial2. La retención de las LDL en el interior de la pared va a favorecer su oxidación, provocando un aumento de la expresión de moléculas de adhesión y proteínas proinflamatorias que estimula la migración de los leucocitos hacia la capa interna de la arteria denominada túnica íntima. Una vez en la pared arterial, los monocitos se diferencian a macrófagos, donde captarán las LDL-ox y se diferenciarán a células espumosas cargadas de lípidos. La formación de la placa de ateroma también implica la actuación de las células de músculo liso vascular (CML) de la túnica media, las cuales migrarán hacia la túnica íntima. En el caso de las arterias humanas, la capa íntima también contiene CML residentes que proliferarán y, junto con las CML que han migrado desde la media, producen proteínas de matriz extracelular como colágeno intersticial y elastina, que favorecen la formación de la capa fibrosa que recubre la placa. Esta capa normalmente se superpone a las células espumosas, las cuales pueden morir y liberar lípidos que se acumulan extracelularmente. La eliminación ineficaz de las células muertas, un proceso conocido como eferocitosis, puede dar lugar a la acumulación de desechos celulares y lípidos extracelulares formando el núcleo necrótico3.
La placa aterosclerótica generalmente causa manifestaciones clínicas derivadas de la estenosis que produce en la luz del vaso, limitando el flujo y dando lugar a isquemia tisular, o al provocar trombos que pueden interrumpir el flujo sanguíneo localmente. Los trombos surgen a menudo después de la rotura física de la capa fibrosa que expone el material procoagulante del núcleo de la placa a las proteínas de coagulación de la sangre, lo que desencadena la trombosis. Las placas más vulnerables a la rotura suelen ser más delgadas, pobres en colágeno y con pocas CML pero abundantes en macrófagos. Las células inflamatorias pueden acelerar la rotura, ya que sintetizan enzimas colagenolíticas que dan lugar a la degradación del colágeno. Además, estas células son capaces de sintetizar mediadores que inducen la muerte celular de las CML, la mayor fuente de colágeno arterial4. Los macrófagos de la placa también producen el factor tisular procoagulante que hace que el núcleo necrótico sea trombogénico. Cuando la placa de ateroma se rompe y se forma el trombo puede causar un infarto agudo de miocardio, ictus, angina inestable o muerte súbita5.
En esta revisión discutiremos el papel que puede jugar una familia de proteínas, las anexinas, en la homeostasis del colesterol, así como en el remodelado vascular patológico asociado al desarrollo y a la progresión de la lesión aterosclerótica.
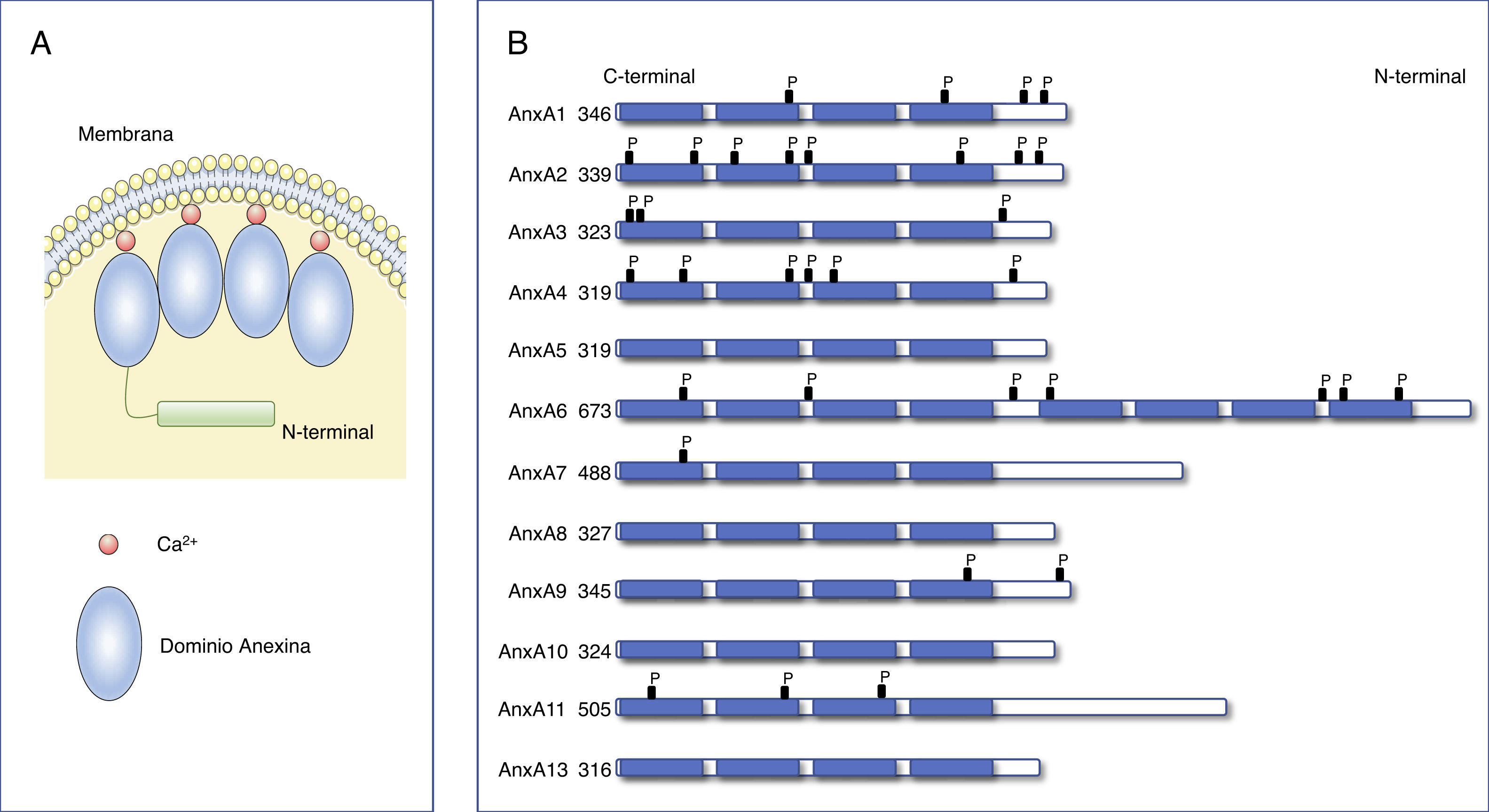
Anexinas. Estructura y función.La familia de las anexinas representa una familia multigénica de proteínas de unión a fosfolípidos dependientes de Ca2+. Se han descrito aproximadamente 100 anexinas distintas en varias especies animales. De ellas, la familia de las anexinas se compone de 12 miembros en humanos y vertebrados referidas como AnxA1-AnxA13 (AnxA12 no está asignado en la actualidad)6-7. El análisis cristalográfico de estas proteínas identificó que todas las anexinas cuentan con dos dominios estructurales: un dominio C-terminal bastante conservado con 4 repeticiones de 70-80 aminoácidos (8 en el caso de la AnxA6) y un dominio N-terminal variable (fig. 1). Cada una de las repeticiones del dominio C-terminal codifica para un sitio de unión a Ca2+, permitiendo que las anexinas se transloquen rápidamente a la membrana plasmática o a membranas intracelulares uniéndose a fosfolípidos cargados negativamente6,8,9. El extremo N-terminal difiere en longitud y secuencia entre las diferentes anexinas y es el responsable de la regulación de la interacción con diferentes ligandos y de la asociación de las anexinas a las membranas8.
 Diagrama de la estructura de las anexinas. Las anexinas se anclan a las membranas a través de la unión a Ca2+. Todas las anexinas contienen un extremo N-terminal variable y un extremo C-terminal conservado compuesto de 4 (8 para la AnxA6) dominios anexina homólogos repetidos que se empaquetan en un compacto y ligeramente disco curvo. (Modificada de Gerke et al.6). B) Estructura primaria de las anexinas humanas. Las anexinas constan de 4 dominios anexina repetidos y un dominio N-terminal variable. El número de la izquierda indica el número de aminoácidos de cada anexina. Las P representan sitios susceptibles de fosforilación.")
Estructura de las anexinas.
A) Diagrama de la estructura de las anexinas. Las anexinas se anclan a las membranas a través de la unión a Ca2+. Todas las anexinas contienen un extremo N-terminal variable y un extremo C-terminal conservado compuesto de 4 (8 para la AnxA6) dominios anexina homólogos repetidos que se empaquetan en un compacto y ligeramente disco curvo. (Modificada de Gerke et al.6).
B) Estructura primaria de las anexinas humanas. Las anexinas constan de 4 dominios anexina repetidos y un dominio N-terminal variable. El número de la izquierda indica el número de aminoácidos de cada anexina. Las P representan sitios susceptibles de fosforilación.
Diferentes estudios in vitro con anexinas purificadas han podido analizar sus propiedades bioquímicas, su plegamiento y su estructura tridimensional, así como sus características de unión a membrana. De esta manera también se han podido determinar sus afinidades por los diferentes fosfolípidos de membrana y con otras proteínas, particularmente de la familia de las proteínas S10010. Las funciones biológicas que desempeñan las anexinas son variadas. La mayoría se desempeñan en el citoplasma celular y su función está íntimamente relacionada con su capacidad de unión a la membrana. Estas proteínas son responsables de los eventos endocíticos y exocíticos regulados por Ca2+ junto con su capacidad para favorecer la estabilización de las membranas de orgánulos y la membrana plasmática11. Uno de las funciones principales de las anexinas es actuar como proteínas de andamiaje a través de la unión regulada por calcio a los fosfolípidos de las membranas. Esto permite que el citoplasma y el lado citoplasmático de la membrana celular interactúen de manera ordenada12. La movilización del calcio intracelular provoca que las anexinas sean reclutadas por las membranas celulares. Sin embargo, algunas anexinas pueden unirse a las membranas en ausencia también de calcio, como las anexinas A9 y A108. Algunos miembros de las anexinas pueden interactuar específicamente con ciertos sitios de ensamblaje de actina en las membranas celulares. Por ejemplo, la organización de microdominios de las membranas celulares de CML está regulada por las anexinas A2 y A6 a través de interacciones con el citoesqueleto13.
Aunque la mayoría de las acciones de las anexinas tienen lugar en el citoplasma celular, bajo determinadas circunstancias algunas anexinas (AnxA2 y AnxA11) también pueden interactuar con el núcleo celular14. En particular, la AnxA11 juega un papel determinante en la fase final de la citocinesis, proceso por el cual se produce la separación física del citoplasma en dos células hijas durante la división celular. Sin este proceso las células no pueden formar el cuerpo medio y entran en apoptosis15. Por otro lado, algunas anexinas han demostrado tener capacidad para estar presentes en la superficie celular. Así, en células incubadas en presencia de glucocorticoides la AnxA1 se transloca del citoplasma a la superficie celular16. También, como veremos posteriormente, la AnxA2 es un correceptor para el plasminógeno en diferentes estirpes celulares, incluidos los macrófagos y las células endoteliales17. Algunas anexinas también se secretan al exterior celular. Así, se ha demostrado que la AnxA5 tiene capacidad anticoagulante y la AnxA1 tiene propiedades antiinflamatorias sobre leucocitos18. Esta última, la AnxA1, se encuentra en el suero humano, y su concentración aumenta principalmente en procesos inflamatorios como la colitis19. Diferentes estudios han demostrado que la AnxA1 inhibe la migración transendotelial de leucocitos, disminuyendo la respuesta inflamatoria20.
La importancia y la participación de las diferentes anexinas en diferentes patologías ha quedado de manifiesto desde el principio de los años 2000, donde se han ido generando animales deficientes en diferentes anexinas como la AnxA1, A2, A4, A5, A6, A7 y A821. Todos los animales deficientes en las diferentes anexinas son viables a excepción de la AnxA7, de la cual se han descrito dos fenotipos diferentes: primero uno letal debido a hemorragia cerebral22 y posteriormente otro normal y viable23. El hecho de que la mayoría de los ratones deficientes en anexinas sean normales y viables sugiere una redundancia en la función de las anexinas. Sin embargo, al analizar las diferentes cepas de animales deficientes y bajo condiciones patológicas revelan resultados distintos identificando mecanismos específicos para cada una de las anexinas. Así, diferencias sutiles observadas en su localización espaciotemporal, diferencias en cuanto a su cinética de unión a fosfolípidos cargados negativamente, diversidad en su interacción por el dominio N-terminal, afinidad por otros lípidos incluidos fosfatidilinositol-4,5-bisfofato, colesterol y ceramidas, modificaciones postraslacionales y, más relevante aún, patrones de expresión diferencial, hacen que haya más oportunidades para describir una diversidad funcional en la familia de las anexinas6.
Anexinas y homeostasis del colesterolUna de las principales funciones de las diferentes anexinas es el control de la homeostasis del colesterol intracelular. El colesterol es un esterol indispensable para la vida que desempeña funciones estructurales y metabólicas. Se encuentra principalmente anclado a las membranas, donde modula su función24. El colesterol es esencial para el crecimiento tisular así como para la síntesis de hormonas esteroideas, ácidos biliares y vitaminaD25. Por lo tanto, la regulación de la homeostasis del colesterol es esencial para la función celular. Como es conocido, aunque el colesterol puede ser sintetizado por las células, la mayor parte del colesterol celular es adquirido a través de la endocitosis de las LDL. Después de la internalización, el colesterol LDL esterificado llega a las endosomas tardíos (ET), donde se hidroliza para ser distribuido en el interior celular a través de proteínas Niemann-Pick C1 y C2 (NPC1/2)26-27. Hasta el 30% del colesterolLDL pasa al retículo endoplásmico (RE) para regular el control de la biosíntesis del colesterol por la propia célula en forma de retroalimentación negativa27-28. Desde el RE, el colesterol puede ser enviado a otros orgánulos, como la membrana plasmática o las mitocondrias. Alternativamente, el exceso de colesterol puede esterificarse mediante acil-coenzima A:colesterol aciltransferasa (ACAT) para su almacenamiento en gotas lipídicas29. El procesamiento del colesterol tiene lugar en diferentes localizaciones subcelulares, transfiriéndose entre las distintas membranas subcelulares por transporte vesicular y por mecanismos no vesiculares27. Por lo tanto, la homeostasis del colesterol debe ser estrictamente regulada para que se mantengan los niveles de colesterol en rangos adecuados y efectúe correctamente sus funciones biológicas.
Recientemente se ha descrito que las anexinas pueden interaccionar directamente con el colesterol regulando su homeostasis30. Por ejemplo, la AnxA1 tiene la capacidad de establecer contactos entre el RE y los cuerpos multivesiculares (CMV) dando lugar a una disminución de la expresión del receptor del factor de crecimiento epidérmico y favoreciendo la transferencia del colesterol desde el RE a los CMV. Esta es una ruta poco usual de transporte de colesterol, ya que las células obtienen colesterol principalmente mediante la endocitosis de LDL y, como se ha comentado, el colesterol derivado de las LDL pasa al RE, regulando a la baja la síntesis de novo de colesterol26-27. Sin embargo, cuando los niveles de colesterol son bajos en los CMV, la ruta se invierte pasando el colesterol del RE a los ET para asegurar la presencia de colesterol para la formación de vesículas en el interior celular31.
En el caso de la AnxA2 se ha observado una asociación de esta anexina con los ET ricos en colesterol32 que podría estar relacionada con el transporte de moléculas desde endosomas tempranos a ET. Cuando hay una disminución de la expresión de AnxA2, aumenta el transporte de moléculas hacia los ET, sugiriendo un papel de la AnxA2 dependiente de los niveles de colesterol32.
Una de las anexinas más importantes en el control de la distribución de colesterol en los distintos compartimentos intracelulares parece ser la AnxA628. La sobreexpresión de AnxA6 se ha relacionado con un aumento de esta anexina en ET que se asocia a un aumento de colesterol dentro de los endosomas y a una disminución de los niveles de colesterol en la membrana plasmática, en el aparato de Golgi y en los endosomas de reciclaje26,33. Los cambios producidos por la AnxA6 en la distribución celular del colesterol interfieren con el tráfico de proteínas SNARE dependientes de colesterol e integrinas, comprometiendo funciones celulares críticas como la adhesión y la migración celular34. Las proteínas SNARE son proteínas sensibles a los niveles de colesterol presentes en membranas de fusión y vesículas de transporte. Los niveles de colesterol en los compartimentos endocíticos regulan la función y la localización de estas proteínas26,35. Además, el colesterol interacciona directamente con algunas proteínas SNARE36-37. Por lo tanto, las anexinas, en concreto AnxA2 y AnxA6, además de regular la fusión de membranas vesiculares mediado por proteínas SNARE y el destino específico de la carga de estas vesículas, también regulan el transporte de colesterol entre diferentes compartimentos celulares.
Los mecanismos moleculares por los que la unión de AnxA6 a los ET altera el transporte de colesterol no están analizados completamente. Aunque las proteínas NPC1/2 son fundamentales para la salida del colesterol de los ET, otras proteínas pueden estar implicadas en el paso de colesterol desde ET a RE. En los ET, proteínas de membrana como NCP1, ORPL1, StARD3 y StARD3NL parecen fundamentales para el paso de colesterol al RE38. Además, la activación de Rab7, una proteínaG pequeña, promueve la movilidad y el reposicionamiento de los ET para la correcta transferencia del colesterol39. En esta movilidad también hay que tener en cuenta la unión de la AnxA6 a las membranas, lo que provoca la activación del reordenamiento de los microdominios ricos en colesterol, como ocurre en la membrana plasmática40. Estos cambios estructurales inducidos por la AnxA6 pueden ocurrir también el los ET, modulando la distribución de colesterol y, en consecuencia, otros lípidos de membrana41. Estos dominios altamente ordenados y ricos en colesterol influyen en el transporte activo del colesterol hacia el RE.
Finalmente, además de la AnxA6, la AnxA8 también controla la homeostasis de colesterol asociada a ET42. Opuesto al papel que desempeña el aumento de la expresión de la AnxA6 en la acumulación de colesterol en ET, la disminución en la expresión de AnxA8 causa el bloqueo de la salida de colesterol de los ET, sugiriendo un equilibrio en la expresión de estas dos anexinas en el control de la carga de colesterol en el interior de los ET42. Por lo tanto, diferentes anexinas parecen jugar un papel importante en el transporte de colesterol a lo largo de la vía endocítica, y cambios en su expresión pueden dar lugar a cambios en la cantidad de colesterol intracelular. Además, el aumento de las concentraciones circulantes de colesterol —y por lo tanto la mayor entrada y acumulación de colesterol en el interior celular— podría ser regulado, al menos en parte, mediante la regulación de la expresión de diferentes anexinas.
Anexinas y remodelado vascularEn los últimos años se ha puesto de manifiesto el papel de diferentes anexinas en la respuesta inflamatoria y en el remodelado vascular subyacente que tiene lugar durante el desarrollo y progresión de la lesión aterosclerótica. Entre las más destacadas se encuentran la anexina A1, A2, A3, A5 y A7. El papel que podrían desempeñar otras anexinas se desconoce en la actualidad.
Anexina A1La AnxA1, también reconocida como lipocortina1, se describió por primera vez cuando se estaban identificando los factores que mediaban acciones antiinflamatorias de los glucocorticoides43-44. Se expresa en la mayoría de tipos celulares y de tejidos, aunque es particularmente abundante en macrófagos, neutrófilos, sistema nervioso y endocrino6,21,45. Como muchas otras anexinas, la AnxA1 se encuentra en diferentes localizaciones celulares, incluyendo membrana plasmática, aparato endosomal, vesículas secretoras, citoesqueleto y núcleo. En estas localizaciones la AnxA1 participa en procesos celulares como transporte de membrana (endo/exocitosis), traducción de señales, dinámica de actina y regulación de enzimas metabólicas relacionadas con proliferación, diferenciación, migración y apoptosis6,8,9,45-47. AnxA1 también desempeña una función extracelular importante, actuando como molécula antiinflamatoria a través de su regulación por glucocorticoides y mediante su unión al receptor de membrana acoplado a proteínasG conocido como receptor2 de péptido N-formílico (formyl peptide receptor 2 [FPR2])48.
El desarrollo del ratón deficiente en AnxA1 puso de manifiesto el papel antiinflamatorio de esta proteína y cómo su ausencia provocaba una respuesta inflamatoria exacerbada y prolongada con un incremento en migración leucocitaria y una resistencia al efecto antiinflamatorio de los glucocorticoides49. De hecho, se ha observado un aumento en los niveles de proteínas proinflamatorias como la ciclooxigenasa2 o la fosfolipasaA2 en estos ratones49. Además, ratones deficientes en su receptor FPR2 mostraron que AnxA1 ejerce su función mediante la activación de la respuesta inmune innata50. Los mecanismos moleculares que subyacen a la activación de FPR2 por AnxA1 se han relacionado con la activación de quinasas activadas por mitógeno (MAPK), como Erk1/2, p38MAPK, Akt,o c-Jun51-52. La activación de estas cascadas de señalización se traduce en una disminución de la adhesión de neutrófilos al endotelio vascular49, incremento de la apoptosis de neutrófilos por vías apoptóticas constitutivas53 o por disminución de señales de supervivencia inducida por otros mediadores inflamatorios54 e inducción de reclutamiento de monocitos y su posterior polarización hacia un fenotipo antiinflamatorio55. Todas estas funciones descritas para la AnxA1 son muy importantes en el desarrollo de la lesión aterosclerótica. Por ello, diferentes estudios han puesto de manifiesto su potencial terapéutico y su capacidad para limitar la formación de la lesión aterosclerótica y las complicaciones cardiovasculares derivadas de ella56. En este sentido, en ratones deficientes en el receptor de LDL (LDLR) alimentados con dieta grasa se observó que la administración intraperitoneal de AnxA1 humana recombinante reducía significativamente la adhesión y el rodamiento de neutrófilos dependiente de FPR2 sobre células endoteliales, atenuando por tanto la progresión de la placa aterosclerótica57. Asimismo, en animales deficientes para AnxA1 o FPR2 en fondo genético ApoE−/− se ha observado un aumento en el desarrollo de lesiones ateroscleróticas asociado a un incremento en la adhesión de células mieloides en las paredes arteriales dañadas, así como aumento en la proliferación de macrófagos dentro de la lesión56,58.
Terapéuticamente, la administración in vivo del péptido Ac2-26 (derivado del dominio N-terminal de la proteína de AnxA1) redujo el reclutamiento de células mieloides dependiente de FPR2, disminuyendo el tamaño de la lesión y la acumulación de macrófagos en las lesiones ateroscleróticas51. En este sentido, el eje AnxA1/FPR2 inhibe la expresión de quimiocinas dependientes de integrinas necesarias para el reclutamiento leucocitario51. Además, en un modelo de aterosclerosis avanzada en ratones deficientes para ApoE tratados con nanopartículas dirigidas específicamente a colágenoIV (ColIV) que contenían Ac2-26 (ColIV-Ac2-26 NP), se observó un aumento del espesor de la cápsula fibrosa, una reducción de la producción de colagenasa y, en consecuencia, una mayor estabilidad de la lesiones ateroscleróticas59. Estos datos indican que la administración de péptidos derivados de AnxA1 podría ofrecer formas farmacológicas alternativas y eficaces para el tratamiento de enfermedades inflamatorias crónicas contrarrestando el reclutamiento continuo de leucocitos y la actividad de macrófagos durante la progresión aterosclerótica.
La importancia de la regulación de la expresión de la AnxA1 viene dada por los diferentes estudios realizados en muestras humanas. Así, AnxA1 se expresa en placas ateroscleróticas de arterias coronarias humanas, colocalizando con macrófagos y células endoteliales y en placas de pacientes con estenosis carotídea sometidos a endarterectomía carotídea60-62. La expresión de AnxA1 fue mayor en placas carotídeas de pacientes asintomáticos comparado con pacientes con sintomatología neurológica, lo que indica un papel protector de AnxA1 en la aterosclerosis60. Además, AnxA1 se expresa principalmente en áreas con alto contenido de células apoptóticas, lo que refleja su importancia en la eliminación de este tipo de células61.
Anexina A2La AnxA2 se expresa en una amplia variedad de tipos celulares, siendo diferente dependiendo del tejido analizado. Así, su expresión es elevada en pulmón y riñón y, sin embargo, reducida en hígado63. AnxA2 se encuentra de forma predominante en la membrana plasmática, en el aparato endosomal y en vesículas secretoras, y solo en algunos casos la encontramos en el núcleo celular64-65. Como otras anexinas, la AnxA2 regula el tráfico y la organización de membranas a través de procesos de endo/exocitosis, formación de microdominios o reparación de la membrana plasmática. Asimismo, su interacción con los filamentos de actina del citoesqueleto celular ha implicado a la AnxA2 en diferentes funciones celulares como crecimiento celular, diferenciación, apoptosis y migración6,9,46,47,65.
La mayoría de AnxA2 se encuentra formando heterotetrámeros con la proteína p11 (S100A10), un miembro de las familia de las proteínas S100. La interacción de AnxA2/p11 actúa como receptor para plasminógeno y activador tisular del plasminógeno (tPA) en células endoteliales, ambas moléculas responsables de promover la fibrinólisis vascular64,66,67. Además, se ha demostrado que la señalización de plasmina/plasminógeno en monocitos humanos utiliza el heterotetrámero de AnxA2 como receptor y desencadena la señalización a través de JAK/STAT, activación de NF-κB dependiente de Akt, así como ERK1/2 y p38 MAPK, lo que conduce a la inducción de genes proinflamatorios y al reclutamiento de células inflamatorias en el contexto de aterosclerosis68.
El desarrollo del ratón deficiente en AnxA2 demostró que estos animales son viables y fértiles, pero muestran una importante deposición de fibrina en algunos tejidos69. En un modelo de trombosis aguda en la arteria carótida en animales deficientes para AnxA2 o para p11 se observó que estos animales mostraban un aumento en la trombosis, lo que demostró que la unión de AnxA2/p11 está implicada en la fibrinólisis vascular70.
El papel que desempeña AnxA2/p11 y sus consecuencias en la homeostasis vascular se han puesto de manifiesto en diferentes modelos experimentales. Por un lado, se ha observado que la inducción de hiperhomocisteinemia mediante la administración de dieta provoca una acumulación de fibrina y alteraciones en la neoangiogénesis en ratones deficientes en AnxA271. Además, en la enfermedad cerebral isquémica la administración de AnxA2 recombinante debe servir como adyuvante para amplificar la trombólisis mediada por tPA y prevenir el ictus72. Sin embargo, la deficiencia de AnxA2 no tuvo ningún efecto en el tamaño de la lesión aterosclerótica en ratones deficientes para ApoE, aunque previamente se hubiera demostrado que la inhibición terapéutica de la generación de plasminógeno es beneficiosa para la aterosclerosis64. En este sentido, la administración de AnxA2 recombinante disminuyó la formación de trombos en un modelo de trombosis en la arteria carótida de rata73. Estos resultados sugieren que la mejora de la actividad fibrinolítica inducida por la AnxA2 podría modular el estado de hipercoagulabilidad en la aterosclerosis.
Como hemos comentado, una de las principales funciones de la AnxA2 está relacionada con la trombosis. Sin embargo, se ha demostrado la implicación de esta proteína en otras respuestas celulares. Así, en ratones deficientes para ApoE alimentados con una dieta rica en colesterol se observó que la expresión de la AnxA2 estaba significativamente aumentada en macrófagos presentes en las lesiones ateroscleróticas74. Esto provocaba un aumento en la migración de macrófagos a través de la señalización por Akt/NF-κB y ERK dependiente de AnxA274. Asimismo, la interacción de la AnxA2 con la integrina α5 a través de la proteína tirosina fosfatasa 1B participa en la respuesta inflamatoria asociada al desarrollo de aterosclerosis en ratones deficientes para ApoE75. Además, AnxA2 se ha implicado en procesos de migración de CML. En este sentido, ensayos de migración in vitro de CML de rata han demostrado que la sobreexpresión o inhibición de AnxA2 aumenta o disminuye, respectivamente, la migración de estas células ante factores de crecimiento76. En consonancia, la expresión de AnxA2 está aumentada durante la formación de la neoíntima en la carótida después de daño por balón76.
Otra de las características importantes de la AnxA2 es su capacidad de interaccionar con la proproteína convertasa subtilisina/kexina tipo NF-κB9 (PCSK9), un potente inductor de la degradación del receptor de LDL hepático63,77. AnxA2 o AnxA2/p11 inhibe la disminución del receptor de LDL mediada por PCSK9 en células HuH7 o HepG277. Además, ratones deficientes en AnxA2 tienen niveles elevados de LDL y de PCSK963. Por lo tanto, la modulación de los niveles de AnxA2 se podría considerar como un inhibidor endógeno de PCSK9, hecho relevante ya que el tratamiento de la hipercolesterolemia se ha beneficiado del uso de anticuerpos monoclonales frente a PCSK9. En los últimos años también se han identificado variantes en el gen de AnxA2 mediante análisis de polimorfismos de un solo nucleótido o SNP que influyen directamente en los niveles circulantes de LDL indicando, de nuevo, a la AnxA2 como una potencial diana terapéutica para la reducción de las concentraciones de LDL78.
La AnxA2 también está relacionada con el metabolismo de la glucosa y de los ácidos grasos. En este sentido, se ha demostrado que la AnxA2 es la responsable de la translocación de GLUT4 inducible por insulina, el principal transportador de glucosa desde los compartimentos intracelulares hasta la superficie celular en adipocitos79. Además, en las células endoteliales y adipocitos del tejido adiposo blanco AnxA2 es fundamental para la captación celular de ácidos grasos a través de su unión al transportador de ácidos grasos CD3679. En estudios in vivo se observó que los animales deficientes en AnxA2 mostraban un claro retraso en el aclarado de ácidos grasos, lo que indica que la falta de AnxA2 comprometía la eliminación mediada por CD36 de ácidos grasos del torrente sanguíneo80. Sin embargo, este mismo estudio demostró que los ratones deficientes en AnxA2 presentaban niveles estables de glucosa así como una tolerancia normal, indicando que AnxA2 no está implicada en la translocación de GLUT4 in vivo80.
Anexina A3Como otros miembros de la familia, la AnxA3 se expresa en una amplia variedad de tejidos, como corazón, pulmón, riñón, cerebro, hígado y, en mayor medida, en tejido adiposo81. Esta proteína está relacionada con diferentes funciones celulares, como la diferenciación y migración celular, la regulación inmune y la formación ósea6. A nivel experimental se ha demostrado que la AnxA3 está implicada en la migración de células endoteliales y en la formación de tubos82. Además, se ha demostrado que el VEGF aumenta la expresión de AnxA3 en células HUVEC, incrementando su migración en ensayos de reparación de herida y la angiogénesis83.
Aunque el ratón deficiente para AnxA3 es viable, la falta de modelos animales usando ratones deficientes para esta proteína no ha permitido hasta el momento analizar el papel que juega en el desarrollo de la lesión aterosclerótica. Sin embargo, la inhibición in vivo de su expresión mediante el uso de un sh-RNA (short hairpain RNA) en un modelo de infarto de miocardio en ratas provocó una disminución en la respuesta inflamatoria, el tamaño del infarto, la expresión de colágenoI yII, favoreciendo la reparación del tejido miocárdico84. Además, mediante la inhibición condicional en el endotelio vascular de ratón (AnxA3f/f;Tie2-Cre) se ha demostrado que la pérdida de AnxA3 no causa defectos en el desarrollo de la vasculatura pero sí es necesaria para el alineamiento paralelo de arterias y venas, imprescindible para un flujo sanguíneo y una función adecuada de los vasos85.
Anexina A5La AnxA5 es el miembro más abundante y más estudiado de la familia de las anexinas. Se expresa en la mayoría de las células y tejidos a excepción de neuronas6,86. Esta anexina ha sido asociada al tráfico de membranas, a la regulación de las canales iónicos y entrada de Ca2+ y, más importante aún, a la regulación del ciclo celular y la apoptosis. Es por eso que la AnxA5 se ha convertido, durante mucho tiempo, en una herramienta de diagnóstico para detectar muerte celular, ya que se une a la fosfatidilserina en la parte externa de la membrana plasmática en células apoptóticas. Además, la AnxA5 tiene otras funciones extracelulares relacionadas con la coagulación, la fagocitosis, la infección viral o los procesos de reparación de la membrana celular86-88.
En cuanto a su relación con la vasculatura, la AnxA5 tiene propiedades antitrombóticas, antiapoptóticas y antiinflamatorias a través de la unión a la fosfatidilserina expresada en la superficie celular89-90. Además, está asociada al síndrome antifosfolípido durante la gestación18,91. Estudios en ratones deficientes en AnxA5 demuestran que las hembras gestantes son más propensas a la pérdida de embriones y, por lo tanto, a camadas más pequeñas92. La administración de anticoagulantes, como la heparina, previene la formación de trombos y, por tanto, la pérdida de embriones durante la gestación en estos ratones.
Asimismo, existen evidencias del papel que desempeña la AnxA5 durante procesos de remodelado vascular. El tratamiento con AnxA5 recombinante redujo la adhesión y la infiltración de leucocitos en placas ateroscleróticas presentes en ratones deficientes para ApoE89. También, en un modelo de daño femoral en ratones ApoE−/− se observó que el tratamiento con AnxA5 disminuía de forma dosis-dependiente la adhesión temprana de leucocitos y macrófagos, y a largo plazo disminuía el desarrollo de aterosclerosis93. Estudios posteriores centrados en los efectos de la administración de AnxA5 exógena demostraron que el tratamiento con esta proteína reduce la inflamación en la aterosclerosis avanzada y contribuye a la estabilización de las placas ateroscleróticas en ratones deficientes para ApoE, sin modificar el tamaño de las lesiones, el contenido de colágeno o de CML90. Sin embargo, el pretratamiento con AnxA5 en un modelo de aterosclerosis temprana en ApoE−/− sí fue capaz de reducir la formación y el tamaño de las placas, disminuyendo la tasa de apoptosis y regulando la infiltración y la activación de macrófagos94. Estos resultados podrían indicar que la AnxA5 juega un papel importante en las etapas iniciales del desarrollo de la lesión aterosclerótica, siendo su papel menos relevante en estadios avanzados. Estudios in vitro refuerzan la idea del papel de la AnxA5 sobre el reclutamiento de células inflamatorias. Así, la AnxA5 exhibió efectos antiinflamatorios en macrófagos reduciendo significativamente el rodamiento, la adhesión y la transmigración de células mononucleares sobre células endoteliales activadas por TNF-α90. Asimismo, la AnxA5 inhibe los efectos proaterogénicos y proinflamatorios de la lisofosfatidilcolina (LPC) en macrófagos in vitro, interfiriendo en la unión y la captación de LDL oxidadas así como en los propios efectos proinflamatorios de la LPC95.
La AnxA5 también se ha propuesto como biomarcador de aterosclerosis en diferentes estudios. Se ha descrito como biomarcador diagnóstico de aterosclerosis subclínica en pacientes con lupus eritematoso sistémico96. Los niveles circulantes de AnxA5 también se han asociado inversamente a la severidad de la estenosis coronaria e incluso se ha observado que la relación oxLDL/AnxA5 es un mejor marcador de la severidad de la estenosis coronaria que la concentración de LDL por sí sola97. En este mismo sentido, varios SNP en el gen de AnxA5 se asocian al riesgo de sufrir reestenosis en pacientes sometidos a angioplastia coronaria93. Finalmente, la AnxA5 se ha usado como biomarcador en estudios de imagen para visualizar inflamación y estrés celular98.
Anexina A7Dentro de la familia de las anexinas, la AnxA7 es la única que presenta dos isoformas: una de 47KDa, que se expresa básicamente en todos los tejidos a excepción del músculo esquelético, y otra isoforma, más grande, de 51KDa, que se encuentra en tejido cardiaco, cerebro y miotúbulos99. Sus funciones se relacionan con la homeostasis de Ca2+, tiene actividad GTPasa, e interviene en la producción de prostaglandinas, en procesos de remodelado cardiaco y en miopatías inflamatorias100. Se han descrito dos fenotipos diferentes para los ratones deficientes en AnxA7. Primero se describió un fenotipo letal debido a hemorragia cerebral22 y posteriormente se demostró que el ratón deficiente para AnxA7 era normal y viable23. El aislamiento y el posterior análisis de sus cardiomiocitos adultos identificaron que estos presentaban frecuencias disminuidas en la contracción celular, lo que indicaba una alteración en la homeostasis del Ca2+ o en el funcionamiento de la maquinaria responsable de la contracción. La AnxA7 interactúa con dos proteínas (sorcina y RyR) responsables del acoplamiento de los canales de Ca2+ a la maquinaria contráctil del músculo cardiaco23,101. Asimismo, se ha demostrado que los ratones deficientes en AnxA7 presentan elevada susceptibilidad a la fibrilación auricular, taquicardia ventricular, arritmia y afectación de la remodelación cardiaca100. En un modelo de constricción aórtica transversal en estos ratones se observó un aumento pronunciado del tamaño de los corazones asociado a un aumento de la expresión de varios genes de hipertrofia, posiblemente inducidos por el factor nuclear cardiaco regulado por Ca2+ de célulasT activadas (NFAT) de una manera dependiente de AnxA7100. Además, la AnxA7 es un regulador endógeno de gen Homeobox1 (HMBOX1) esencial para la supervivencia de las células endoteliales. Así, niveles disminuidos de AnxA7 debido al tratamiento con su inhibidor 6-amino-2,3-dihidro-3-hidroximetil-1,4-benzoxazina elevaban los niveles de HMBOX1102. En cuanto al remodelado vascular asociado a la formación de la lesión aterosclerótica, se ha demostrado que el tratamiento de ratones deficientes para ApoE con un inhibidor de AnxA7 reduce el área de la placa aterosclerótica, el depósito de lípidos y macrófagos proinflamatorios, así como un aumento de macrófagos antiinflamatorios, contenido de colágeno y CML en las lesiones ateroscleróticas, aumentado la estabilidad de las placas103.
Otras anexinasEn la actualidad se conoce discretamente o se desconoce el papel que juegan otras anexinas sobre el remodelado vascular patológico. En este sentido, únicamente la AnxA10 se ha relacionado con la formación de la lesión aterosclerótica, asociando su expresión con la presencia de placas más estables104, posiblemente relacionada con la actividad anticoagulante, antiinflamatoria o antiapoptótica observada para otras anexinas. Además, la AnxA8 regula el transporte de proteínas por el endosoma tardío en células endoteliales que contribuye al control de la exposición de CD63 hacia el exterior de la membrana plasmática para el correcto reclutamiento de leucocitos105-106. En este sentido, ratones deficientes en AnxA8 presentan menor adhesión y rodamiento leucocitario106.
Finalmente, la AnxA4 tiene 3 transcritos diferentes (AnxA4a, AnxA4b y AnxA4c) que presentaban patrones únicos de expresión107. Aunque no se ha demostrado su relación con la lesión aterosclerótica, su función principal se asocia a la señalización de β-adrenorreceptores (β-AR)/cAMP en cardiomiocitos108. Debido a que los cardiomiocitos de ratones adultos deficientes para AnxA4 mostraron un aumento de los niveles de cAMP, se describió que probablemente era debido a la pérdida de la acción inhibidora de AnxA4 en adenilil ciclasa5, que controla la conversión de ATP en cAMP. Además, se observó que ratones AnxA4−/− tratados con agonistas β-AR mostraban niveles aumentados de AMPc, asociados a una mejora en la fuerza de contracción cardiaca108.
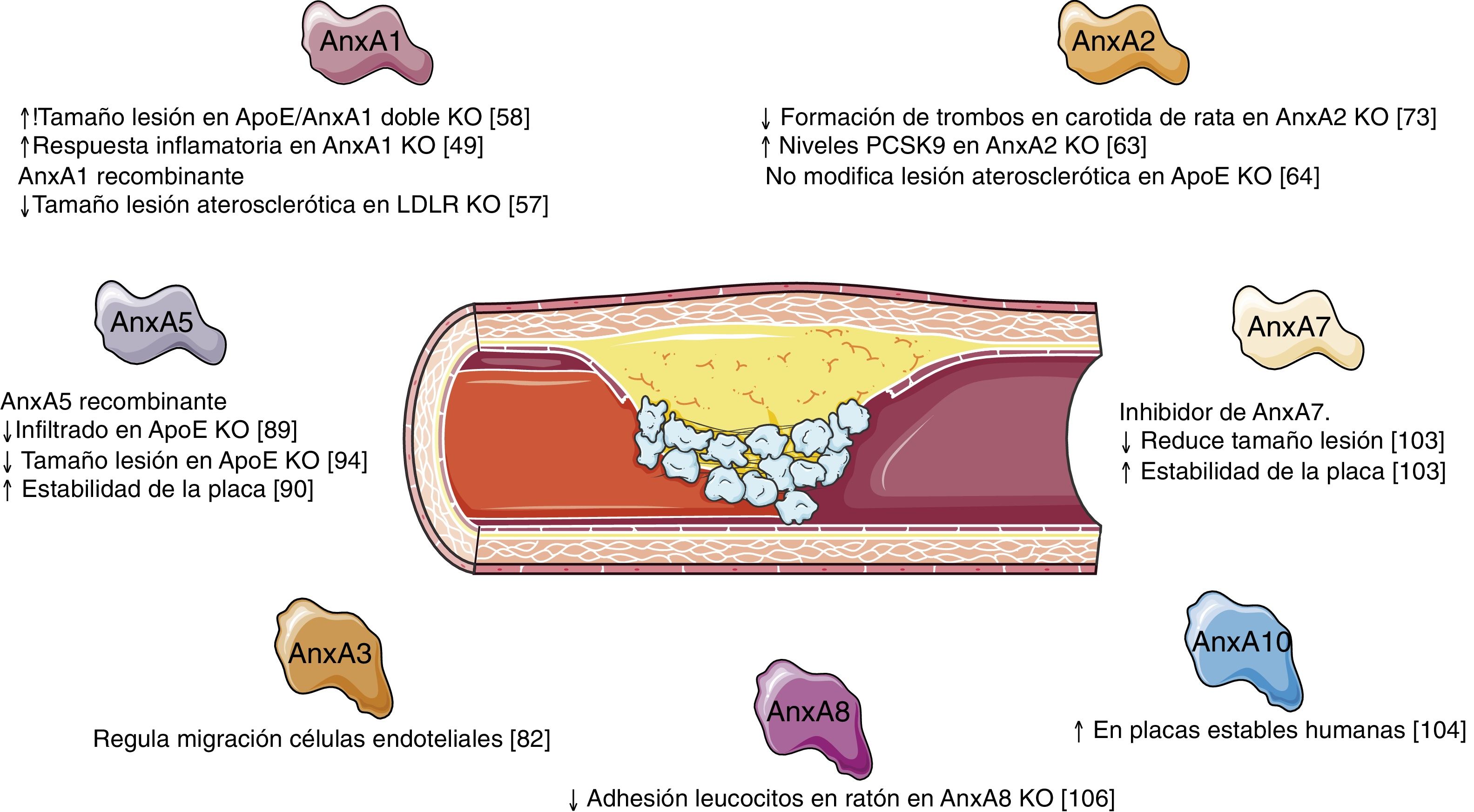
ConclusiónLas anexinas son una familia de proteínas que parecen jugar un papel relevante en la homeostasis del colesterol y en el remodelado vascular que subyace a la formación y el desarrollo de la lesión aterosclerótica (fig. 2). Aunque en los últimos años se ha avanzado en el conocimiento del papel que desempeñan algunos miembros de esta familia, todavía se desconoce qué papel pueden jugar otros, por lo que parece necesario ampliar nuestro conocimiento sobre las acciones de las diferentes anexinas y su posible sobreexpresión/inhibición como opción terapéutica en el tratamiento del remodelado vascular.
Financiación
Este trabajo ha sido realizado gracias a una beca FEA/SEA de investigación básica 2018 de la Sociedad Española de Arteriosclerosis, el Instituto de Salud CarlosIII (ISCIII-FEDER) PI16/01419 y PI19/00128. N. M-B obtuvo un contrato Miguel Servet del ISCIII (CP19/00151).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores piden disculpas a todos aquellos investigadores cuyo trabajo no ha podido ser citado debido a las limitaciones de espacio.