







Familial adenomatous polyposis is described as one of the common two types of genetic disorders: APC and MUTYH gene associated polyposis syndrome and the clinical differences between the two can sometimes be unclear.
Materials and methodsA retrospective analysis and comparison was made of clinical, surgical, and histological criteria, mutation types and the long-term results of patients who underwent genetic analysis which resulted in the diagnosis of familial adenomatous polyposis between 1984 and 2018.
ResultsOf the total 71 patients included in the study, 14 were identified with the MUTYH gene, and 57 with the APC mutation. In patients with the APC mutation, 63% had duodenal adenoma, 61% gastric polyp and 54% had desmoid tumor. Of the patients with the MUTYH mutation, 21% had duodenal adenoma and 21% were diagnosed with gastric polyps. In 21% of the patients with APC mutation, the polyp count was <100, and 64% of those with the MUTYH mutation had >100 polyps in the colon No statistical difference was determined between the groups in respect of the proportion of patients with >100 polyps.
ConclusionThe pre-operative genetic testing of patients with polyposis coli will be useful in determining the future clinical outcome and helpful in guiding an informed decision as to whether to apply surgical treatment. It is useful to determine the colonic and extra-colonic involvement of genetic mutation diseases in patients with familial adenomatous polyposis.
La poliposis adenomatosa familiar (PAF) es una patología hereditaria, caracterizada por la existencia de pólipos y cáncer en el colon. La PAF se describe como uno de los dos tipos más frecuentes de trastornos genéticos: El gen adenomatous polyposis coli (APC) o el gen mutación Y homólogo (MUTYH), genes asociados con el síndrome polipoide. Muchas veces las diferencias clínicas y fenotipicas entre las dos alteraciones geneticas no estan claramente establecidas.
Materiales y métodosSe realizó un analisis restrospectivo de las manifestaciones clinicas, criterios quirurgicos e caracteristicas histologicas, tipo de mutacion y resultados a largo plazo de pacientes diagnosticados mediante analisis genticos de poliposis adenomatosa familiar entre 1984 y 2018.
ResultadosDe un total de 71 pacientes incluidos en el estudio, en 14 de ellos se identificó mutación del gen MUTYH y en 57, mutación del gen APC. A 60 pacientes se les realizó tratamiento quirúrgico, a la mitad de ellos se les practicó proctocolectomía y a la otra mitad, colectomía total. En pacientes con la mutación APC, el 63% presentó; el 61%, y el 54%, tumor desmoide. De los pacientes con la mutación del gen MUTYH, el 21% presentó y al 21% se le diagnosticó pólipos gástricos. En el 21% de los pacientes con mutación del gen APC, el número de pólipos fue inferior a 100 y en el 64% de los pacientes que presentaron mutación del gen MUTYH se observaron más de 100 pólipos en el colon. No existió diferencias estadísticamente significativas entre lo grupos respecto a la proporción de pacientes con más de 100 pólipos.
ConclusiónEs importante valorar la afectación colónica y la extracolónica en pacientes con mutaciones genéticas asociadas a la PAF.
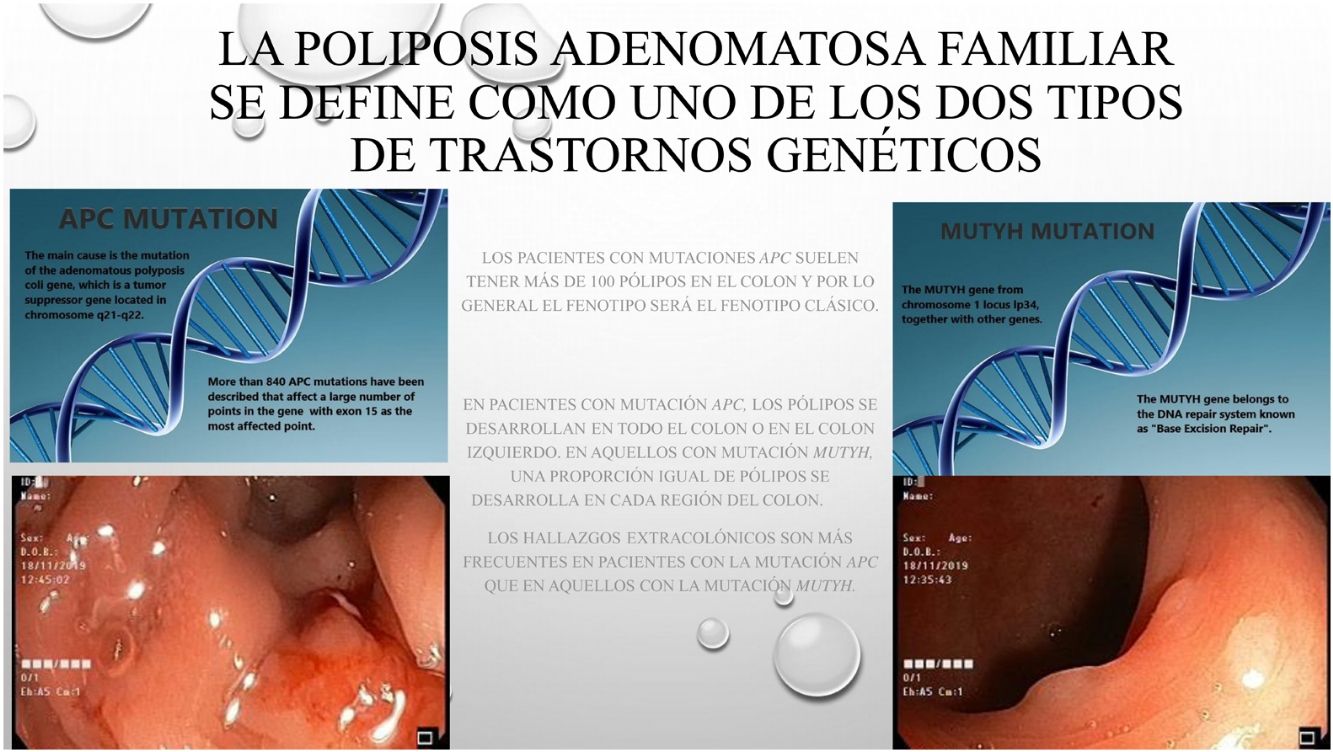
Based on data from the European medical agency, the incidence of familial adenomatous polyposis (FAP) has been reported to be 3–10/10000 of the general population.1,2 The disease has autosomal dominant transmission and these traits can be observed in the family line.3 The main cause is mutation of the adenomatous polyposis coli (APC) gene, which is a tumor suppressor gene located in chromosome 5q21-q22. More than 840 APC mutations have been described that affect a large number of points in the gene with exon 15 as the most affected region.3,4 The mutation determines the location and phenotypic detail of the invasion area. Another rarely observed mutation is the mut Y homolog gene (MUTYH)-associated polyposis mutation (MAP). The MUTYH gene from chromosome 1 locus lp34, together with other genes, belongs to the DNA repair system known as “Base Excision Repair”.5 Genes such as: POLE, POLD1, NTHL1 and MSH3 also contributes to colorectal adenomatous polyp etiology.6,7
The aim of this study was to determine the extension and timing of colon involvement, extracolonic involvement, and the long-term outcome of patients with genetic mutations to be able to better manage the treatment of patients with FAP.
Materials and MethodsA retrospective review was made of patients diagnosed with FAP between 1984 and 2018 on the basis of genetic analysis. The clinical, radiological, operative and pathological results and follow-up data of the identified patients were analyzed and compared. Patients with incomplete or unavailable data were excluded from the study. Diagnosis was done by genetic analysis in all patients. For the cases with positive family history, genetic analysis was used solely without the need of clinical diagnosis. On the other hand the patients who had already been diagnosed clinically were scheduled for genetic test later on.
Genetic TestingDNA extraction was conducted using peripheral blood samples and by sequencing the integrity of the gene if a mutation was not found in the mutation cluster region (codon 1250–1550) of APC. For patients without an APC mutation, a search for the MUTYH mutation was performed as previously described.
According at diagnosis, the patients were divided into two groups: Group 1 comprised patients with APC gene mutation, and Group 2 comprised patients with the MUTYH gene mutation. The 2 groups were compared in respect of age and gender of the patients, the type of surgical colon resection if they had been operated on, operating time, and whether surgery was laparoscopic or (conventional) open surgery.
The number of polyps in the specimen was examined as phenotype and specimens were classified in 2 groups as >100 polyps categorized as classical type and <100 polyps as attenuated type (AFAP). It was determined whether the polyps detected in the patients were denser in the colon. During the study, it was also recorded whether or not they were initially diagnosed with cancer, whether they developed colonic polyps or colon cancer during follow-up after colon resection, whether they had duodenal adenoma, gastric polyp or desmoid tumors (and determination of the timing and duration) and the presence or absence of less common extra-intestinal diseases (retina problems, thyroid cancer, thyroid nodule, adrenal adenoma, breast cancer), whether there was a family history of FAP, their life span and finally their disease-free lifespan.
In the follow-up of patients, colonoscopy, gastroduodenoscopy, and abdomen and thyroid ultrasound examinations were applied annually. Computed tomography (CT) and magnetic resonance imaging (MRI) were also applied together with ophthalmic examination in a systematic way to confirm the diagnosis, and with any other necessary consultations. In suspicious cases these tests were applied more often and the treatment was organized accordingly.
Statistical AnalysisVariables were presented as mean±standard deviation (SD) or median (range, interquartile range [IQR]) values for continuous data and as ordinal data. Categorical variables were analyzed with the Chi-square or Fisher exact test as appropriate. Continuous variables with normal distribution were analyzed with the unpaired t-test. The Kolmogorov–Smirnov test was used to assess the conformity of continuous variables to normal distribution. Survival was analyzed graphically with the Kaplan-Meyer method and comparisons were made using the Log-rank test. A two-sided P value of <.05 was considered statistically significant. Statistical n analysis was performed using commercially available software (IBM-SPSS version 25.0 for Windows; SPSS).
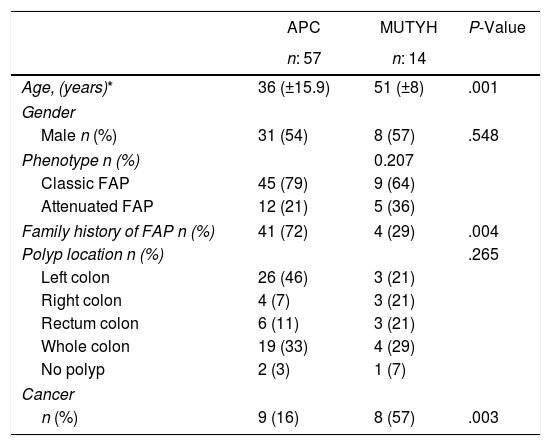
ResultsOf the 71 patients who were diagnosed, 14 (20%) had the MUTYH mutation and 57 (80%) had the APC mutation (Table 1).
Demographic Phenotypic and Clinical Data According to the Genetic Characteristics of the Patients With Familial Adenomatous Polyposis.
| APC | MUTYH | P-Value | |
|---|---|---|---|
| n: 57 | n: 14 | ||
| Age, (years)* | 36 (±15.9) | 51 (±8) | .001 |
| Gender | |||
| Male n (%) | 31 (54) | 8 (57) | .548 |
| Phenotype n (%) | 0.207 | ||
| Classic FAP | 45 (79) | 9 (64) | |
| Attenuated FAP | 12 (21) | 5 (36) | |
| Family history of FAP n (%) | 41 (72) | 4 (29) | .004 |
| Polyp location n (%) | .265 | ||
| Left colon | 26 (46) | 3 (21) | |
| Right colon | 4 (7) | 3 (21) | |
| Rectum colon | 6 (11) | 3 (21) | |
| Whole colon | 19 (33) | 4 (29) | |
| No polyp | 2 (3) | 1 (7) | |
| Cancer | |||
| n (%) | 9 (16) | 8 (57) | .003 |
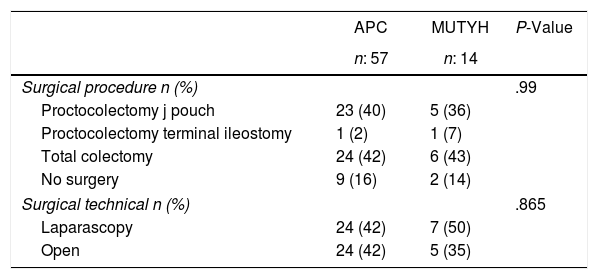
Genetic analysis was applied to 11 patients (APC:9, MUTYH:2) with familial history before the development of colonic polyps and these patients were then followed up with a colonoscopy. APC mutation was diagnosed in patients at the average age of 22.8±10.8 years. Among those cases 2 of them have been diagnosed for extra intestinal involvement (thyroid nodule, thyroid cancer). Of the 60 patients who were operated on, proctocolectomy was applied to 30 (50%) and total colectomy to 30 (50%) (Table 2).
Surgical Procedures According to the Genetic Characteristics of the Patients With Familial Adenomatous Polyposis.
| APC | MUTYH | P-Value | |
|---|---|---|---|
| n: 57 | n: 14 | ||
| Surgical procedure n (%) | .99 | ||
| Proctocolectomy j pouch | 23 (40) | 5 (36) | |
| Proctocolectomy terminal ileostomy | 1 (2) | 1 (7) | |
| Total colectomy | 24 (42) | 6 (43) | |
| No surgery | 9 (16) | 2 (14) | |
| Surgical technical n (%) | .865 | ||
| Laparascopy | 24 (42) | 7 (50) | |
| Open | 24 (42) | 5 (35) | |
APC: adenomatous polyposis coli, MUTYH: Mut Y homolog gene.
Endoscopic ampullectomy was applied to 14 (24.5%) patients. The Whipple procedure was performed in 2 (3.5%) patients with Ampulla of Vater tumors. In the examination of the association between the operation and desmoid tumor, 4 patients were recorded with desmoid tumor before surgery. In the other patients, desmoid tumors developed after an average of 93 (1–246) months, and there were 18 (58%) patients with a previous family history. In 11 (57.9%) patients applied with proctocolectomy and in 12 (60%) applied with total colectomy, a postoperative intra-abdominal desmoid tumor developed. There was determined to be no postoperative development of intra-abdominal desmoid tumor in 8 (42.1%) patients applied with proctocolectomy and in 8 (40%) applied with total colectomy. No statistically significant difference was determined between these groups in respect of the rate of development of postoperative desmoid tumor (P: .894) (Table 3).
Extra-colonic Disease According to the Genetic Characteristics of the Patients With Familial Adenomatous Polyposis.
| APC | MUTYH | P-Value | |
|---|---|---|---|
| n: 57 | n: 14 | ||
| Duodenal adenoma n (%) | 36 (63.2) | 3 (21.4) | .004 |
| Duodenal adenoma age, (years)* | 40 (±13.2) | 55 (±4.1) | |
| Gastric polyp n (%) | 35 (61) | 3(21) | .006 |
| Gastric polyp age, (years)* | 41 (±15.2) | 57 (±9.4) | |
| Desmoid n (%) | 31 (54) | 0 | <.01 |
| Abdominal wall | 5 (16) | 0 | |
| Intra-abdominal | 12 (39) | 0 | |
| Intra-abdominal+Abdominal wall | 14 (45) | 0 | |
| Desmoid age, (years)* | 38.9 (±10.2) | 0 | |
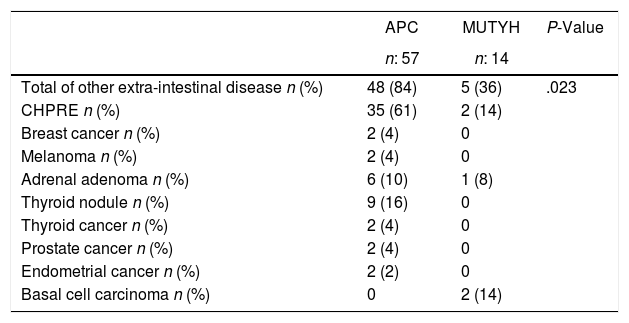
When other extra intestinal diseases were examined, These diseases were rarely detected in patients with MUTYH (Table 4).
Other Extra Colonic Disease According to the Genetic Characteristics of the Patients With Familial Adenomatous Polyposis.
| APC | MUTYH | P-Value | |
|---|---|---|---|
| n: 57 | n: 14 | ||
| Total of other extra-intestinal disease n (%) | 48 (84) | 5 (36) | .023 |
| CHPRE n (%) | 35 (61) | 2 (14) | |
| Breast cancer n (%) | 2 (4) | 0 | |
| Melanoma n (%) | 2 (4) | 0 | |
| Adrenal adenoma n (%) | 6 (10) | 1 (8) | |
| Thyroid nodule n (%) | 9 (16) | 0 | |
| Thyroid cancer n (%) | 2 (4) | 0 | |
| Prostate cancer n (%) | 2 (4) | 0 | |
| Endometrial cancer n (%) | 2 (2) | 0 | |
| Basal cell carcinoma n (%) | 0 | 2 (14) |
CHPRE: Congenital hypertrophy of the retinal pigmented epithelium, APC: adenomatous polyposis coli, MUTYH: Mut Y homolog gene.
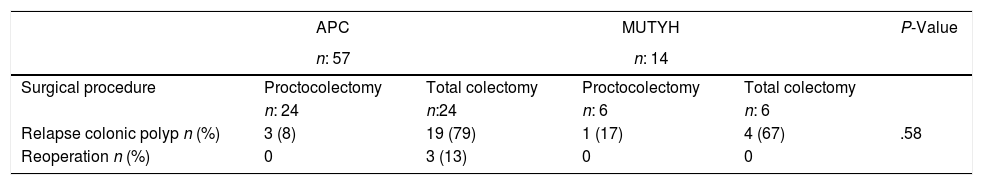
In general, recurrent development of colonic polyps was detected in 23 (79.3%) patients who underwent total colectomy, and follow-up rectoscopic polypectomy was performed. In 19 (73%) patients with APC mutation who also underwent total colectomy, colonic polyps recurred and therefore rectoscopic polypectomy was required during follow-up. The proctocolectomy procedure was performed in 3 (13%) patients, because during follow-up, polyps were detected in the rectum with increased numbers of dysplasia and polyps. No re-operations were applied to patients with MUTYH mutation, who had previously undergone total colectomy and these patients were followed up with polypectomy (Table 5).
Long-term Outcomes According to the Surgical Procedures and the Genetic Characteristics of the Patients With Familial Adenomatous Polyposis.
| APC | MUTYH | P-Value | |||
|---|---|---|---|---|---|
| n: 57 | n: 14 | ||||
| Surgical procedure | Proctocolectomy | Total colectomy | Proctocolectomy | Total colectomy | |
| n: 24 | n:24 | n: 6 | n: 6 | ||
| Relapse colonic polyp n (%) | 3 (8) | 19 (79) | 1 (17) | 4 (67) | .58 |
| Reoperation n (%) | 0 | 3 (13) | 0 | 0 |
APC: adenomatous polyposis coli, MUTYH: Mut Y homolog gene.
The mean duration of follow-up was 17±1.7 years for patients with APC mutation and 19±4.1 years for those with MUTYH mutation (P: .521). During the follow-up of the patients with APC mutation, 34% had no FAP- related complications and 55% had FAP- related complications that had arisen due to the APC mutation. Mortality was seen in 5 patients with APC mutation during the follow-up period, as 1 patient with metastases of colon cancer, 1 patient with desmoid complications, and 3 for other reasons. Of the patients with MUTYH mutation, 57% were disease-free during the follow-up period and 43% were followed up with disease progression. In this group, mortality was seen in 4 patients, as 3 patients with metastasis of colon cancer and 1 for other reasons (Fig. 1, Table 5).
Discussion APC: Adenomatous polyposis coli, MUTYH: Mut Y homolog gene.")
Polyposis coli is one of the most genetically researched diseases. In general, mutations of the APC gene and MUTYH gene have been identified in patients diagnosed with FAP. The objective of this study was to contribute to the better management of patients with FAP through the determination of the extent and timing of colon involvement, extracolonic involvement, and long-term outcomes of patients with genetic mutations.
Patients with APC mutations generally have more than 100 polyps in the colon and in general the phenotype will be the classic phenotype. In the current series, 20% of the patients with APC mutation had a polyp count below 100, and in contrast, more MUTYH mutation patients than expected were detected with more than 100 polyps. Contrary to expectations, no statistical difference was determined between the two groups of patients who had more than 100 polyps. The incidence of >100 colon polyps in the cases with APC mutations4 was similar to that of many previous studies. In general, there were fewer MUTYH mutation patients reported with >100 colon polyps.7,8
When the anatomic features were examined, it was determined that, consistent with literature, in patients with APC mutation, polyps developed in either the whole colon or in the left colon.4 In those with MUTYH mutation, an equal ratio of polyps was determined in each region of the colon. However, in one study, the right colon was reported to be mostly affected in the presence of the MUTYH mutation.9 Both groups were examined and a statistically significantly higher number of patients with MUTYH mutation were diagnosed with tumor diagnosis in the first operation compared to the patients with APC. It was also found that more patients with APC mutation had familial history and were therefore more likely to have undergone familial screenings, resulting in diagnosis without tumor development and a higher incidence of prophylactic colectomy. Similar to previous findings, the current study patients with MUTYH mutations were also seen to be more likely to have been initially diagnosed with tumors.4
When the features of the extracolonic diseases were examined, duodenal adenoma and gastric polyps were found to be statistically significantly more frequent in patients with APC mutation than in MUTYH mutation. The rate of gastric polyp detection was seen to be similar to that of MUTYH mutation cases, but ampulloma has been reported to be more prevalent in literature.10 Ampulloma are also important in these patients after having undergone prophylactic colectomy. Initially benign lesions can progress to cancer with an incidence rate of just under 5%.11 Since this disease is initially a benign condition, it is best to treat these patients with a minimally invasive method. Endoscopic ampullectomy is the most commonly used technique in these patients, as the most appropriate for minimal mortality and morbidity.11,12 Although duodenal adenomas have been reported to progress and develop into pancreatic cancer in these patients, reduced frequency has been observed with the implementation of endoscopic follow-up and subsequent interventions.13 However, the occurrence rate of APC mutation together with duodenal adenoma has been noted as 40%–90%.14 The mean age of detection of duodenal adenomas and gastric polyps appears to be 40 years. The diagnosis of patients with MUTYH mutation at a later age was found to be statistically significant.
Desmoid tumor is the second most common cause of death in patients with polyposis coli and mortality develops in approximately 10%15 of cases. Risk factors for desmoid tumors can be described as a germ-line mutation beyond codon 1444, a family history of desmoid tumor, and a personal history of abdominal surgery.4,16,17 Previous studies have reported that the risk of desmoid tumor development does not change according to the type of operation.18 In a study by Nieuwenhuis et al.19 of 2260 patients, there was determined to be no difference between patients applied with total colectomy and proctolectomy in respect of the development of desmoid tumor. Consistent with the findings in literature, no statistically significant difference was determined in the current study between patients applied with total colectomy and those with proctocolectomy in respect of the development of desmoid tumor. Previous studies15 have reported the lifetime risk of desmoid tumor development in patients with APC mutation to be 10%–30%, and in the current study this was found to be higher at 54%. This can be attributed to the current study patients having a familial history of desmoid tumor at the rate of 58% and could also be associated with the genetic mutation locations. The mean age of appearance of desmoid tumor was determined as 39 years. In the current study, the incidence rate of the diagnosis of the desmoid tumor before performing prophylactic colectomy was 13% and this result was found to be relatively higher than rates reported in previous studies.4,16 Extracolonic diseases were diagnosed in patients with the APC mutation at a statistically significantly higher rate.17
FAP-associated thyroid carcinoma is the third most common cancer type, with a life-time development risk of 1%–2%.20 In the yearly screenings performed in the current study, thyroid ca was determined at the rate of 4%, and thyroid nodules at 16%. Adrenal adenoma may develop at the rate of 7%–13% in patients with FAP,20 and consistent with these reports in literature, adrenal adenoma developed in 10% of the current series. Congenital hypertrophy of the retinal pigmented epithelium (CHPRE) is an age-related pigment fundus lesion, which is a phenotypic marker for FAP with reported prevalence of 70%–75%.21,22 In the current series, this was determined at 61%, a lower rate than previous findings in literature.
In the treatment of patients with prophylactic colectomy, cancer has been observed to develop around the age of 39 years. Therefore, it is better for these patients to undergo early prophylactic colectomy to ensure a good future clinical outcome.13 The goal of prophylactic colectomy is to maintain as high a quality of life as possible and to protect the patient from colorectal cancer.23,24 In recent years, laparoscopic proctocolectomy, mucosectomy and j pouch have been preferred for our patients. In proctocolectomy the quality of life is much lower than that of total colectomy.25 Total colectomy can be performed if the mutation has been checked and preoperative colonoscopy does not determine polyp localization to be intense around the rectum and surroundings. The proctocolectomy was performed before the MUTYH mutation was defined, due to the presence of multiple polyps of uncertian histology, which could not be examined with endoscopy and also due to the risk of the multiple dysplastic polyps degenerating to a rectal cancer.
Despite a greater number of re-operations on total colectomy patients than proctocolectomy patients, no statistically significant difference was found. In the current study population of polyposis patients from a single center, even with the largest mutation definition, we were able to find only 23.9% patients with a suspected AFAP mutation. This low number of patients makes it difficult to reach a precise definition of this syndrome and to determine treatment guidelines. For patients with suspected AFAP, conservation of the rectum is recommended as rectal polyps are thought to occur less frequently.4,21 Care must be taken to closely follow-up the patients and if polyps are observed in the rectum, proctocolectomy may be performed as the second surgical procedure.
The limitations of this study include the retrospective design and that the characteristics of patients were not specified according to the mutation site.
In conclusion, genetic testing of patients with FAP will be useful in determining the future clinical outcome and will be guiding in deciding the type of surgical treatment.
FundingNone.
Conflict of InterestsThe authors declare that they have no conflict of interests.
Please cite this article as: Bademci R, Bollo J, Ramón y Cajal T, Martínez MC, Hernández MP, Targarona EM. Presentación y seguimiento de poliposis adenomatosa familiar (PAF): diferencias entre las mutaciones APC y MUTYH. Cir Esp. 2020;98:465–471.
