







Los materiales cerámicos se están empleando, cada vez en mayor medida, en aplicaciones biomédicas debido a que son capaces de generar una osteointegración completa en el cuerpo humano. Esto se produce debido a que en contacto con sistemas fisiológicos estos materiales experimentan ciertas transformaciones que facilitan la formación de hueso nuevo alrededor del implante cerámico. En este trabajo se pretende llevar a cabo un estudio de bioactividad de un material del sistema P2O5-CaO-SiO2-MgO, para ello se generaron muestras densas de dicho material por fusión zonal por láser (LFZ) y se llevó a cabo el estudio microestructral del mismo, antes y después de llevar a cabo el ensayo de bioactividad, tanto de materiales vítreos como cristalinos.
Partiendo del material en polvo hemos generado cilindros cerámicos por prensado isostático en frío y después de llevar a cabo la sinterización de los cilindros conformados, generamos barras densas mediante la técnica de LFZ. Con el objetivo de controlar la microestructura de las muestras, se lleva a cabo un barrido de la velocidad de crecimiento, siendo capaces de elegir entre una estructura cristalina o vítrea. Las muestras obtenidas son bioactivas en contacto con suero fisiológico artificial ya que son capaces de generar una capa de hidroxiapatita en la superficie del material, cuya función es promover la unión hueso-implante y la regeneración del tejido dañado.
Ceramic materials are increasingly used in biomedical applications due to their ability to produce complete osseointegration in the human body. This is possible because, in contact with physiological systems, these materials undergo certain transformations that facilitate the formation of new bone around the ceramic implant. In this research is intended the bioactivity study of a material of the P2O5-CaO-SiO2-MgO system. For this reason the preparation, by zonal fusion by laser, the microstructural characterization and the study of the in vitro response of a material, both vitreous and crystalline, are proposed.
Starting from the powdered material we have generated ceramic rods by cold isostatic pressing. After the sintering process of the rods at 1200˚C, we have achieved dense bars by laser floating zone technique (LFZ). An exploration of experimental conditions was performed, being able to obtain both, vitreous and crystalline structure. Samples obtained are bioactive in contact with simulated body fluid because they are able of generate an hydroxyapatite layer on the surface of the material whose function is to promote the union of the implant to the bone and the regeneration of damaged tissue.
Los materiales cerámicos se emplean, cada vez más, en aplicaciones biomédicas debido a su capacidad de producir una osteointegración completa en el cuerpo humano [1,2]. Para este fin, es recomendable el uso de estructuras porosas interconectadas; dichos poros permiten la vascularización del implante, así como la colonización del mismo por células, generando tejido óseo nuevo. Esa red porosa idealmente debe estar formada por una combinación de macro y microporos, en la que el 60% de los poros deben tener un tamaño de entre 150 y 400μm y al menos el 20% debe tener un tamaño menor a 20μm [3].
La desventaja de los materiales porosos es que presentan peores propiedades mecánicas que los densos. Para solventar este inconveniente, de Aza et al. [4–6] desarrollaron un material, denominado Bioeutectic® (marca registrada por P.N. De Aza, F. Guitián y S. De Aza), que es capaz de generar in situ una estructura de hidroxiapatita (HA) con porosidad interconectada, al estar en contacto con suero fisiológico artificial (SFA), a la temperatura del cuerpo humano (36,5°C). Dicho material presenta una estructura lamelar eutéctica con 2fases, una de ellas reabsorbible y la otra bioactiva, para que, en el cuerpo humano, se produzca la disolución de una de las 2fases dando lugar a una estructura similar al hueso. La fase que se disuelve es la wollastonita (CaSiO3) y el fosfato tricálcico (TCP) se transforma a HA, dando lugar a una estructura con porosidad interconectada de HA.
En este trabajo se propone estudiar la respuesta en el SFA de un material que además de presentar silicio, como el Bioeutectic®, contiene magnesio, elementos que ejercen un papel muy importante en la regeneración ósea. El silicio desarrolla un papel relevante en los procesos de mineralización ósea alrededor del implante, ya que experimenta una disolución parcial que da lugar a una capa de Si amorfo, involucrada en la regulación de genes, la proliferación y diferenciación de células osteoblásticas, la síntesis de colágeno tipo i y la formación de una capa de HA que favorecería la unión del implante al tejido dañado [7–10]. El magnesio, por su parte, es esencial para el metabolismo humano y se encuentra presente en el tejido óseo [11–16]. Además, está reportado que el magnesio puede tener efectos estimulantes sobre el crecimiento de nuevo tejido óseo [17–20].
La influencia de estos elementos en sistemas modificados a partir del Bioeutectic® ha sido previamente analizada por de Aza et al., llevando a cabo el estudio de bioactividad de materiales con composición Ca3(PO4)2-CaMg(SiO3)2 (TCP-D) [21], CaSiO3-CaMg(SiO3)2 (W-D) [22], y CaSiO3-Ca3(PO4)2-CaMg(SiO3)2 (W-TCP-D) [23]. En este último trabajo, la obtención del material cristalino ternario se llevó a cabo mediante preparación vítrea con posterior desvitrificación, en cambio en nuestro trabajo se persigue la obtención, mediante fusión zonal por láser (LFZ), de un material cuyo ordenamiento, cristalino o vítreo, puede elegirse durante el proceso de fabricación, por el simple hecho de modificar la velocidad de crecimiento durante el proceso de solidificación direccional por láser.
Por este motivo, y debido a la dificultad de obtener estructura cristalina del eutéctico ternario, en este trabajo se plantea la preparación mediante LFZ, la caracterización microestructural y el estudio de bioactividad de un material (M), tanto vítreo como cristalino, del sistema P2O5-CaO-SiO2-MgO, cuya composición (tabla 1) se desvía ligeramente de la composición eutéctica ternaria.
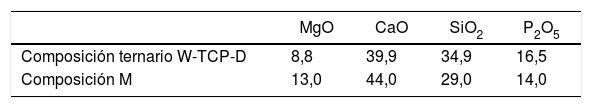
Comparación de las composiciones del eutéctico ternario y del material M (composición desviada del eutéctico CaSiO3-Ca3(PO4)2-CaMgSi2O6), presentada en porcentaje en peso de los óxidos que constituyen las 2composiciones
| MgO | CaO | SiO2 | P2O5 | |
|---|---|---|---|---|
| Composición ternario W-TCP-D | 8,8 | 39,9 | 34,9 | 16,5 |
| Composición M | 13,0 | 44,0 | 29,0 | 14,0 |
Se demuestra que el material obtenido es bioactivo, por lo que podría emplearse como implante óseo al ser capaz de generar in situ una estructura porosa.
MetodologíaLos materiales de partida, SiO2 (pureza 99,8%, Alfa Aesar, Haverhill, Massachusetts, Estados Unidos), silicato de calcio (pureza>99%, Merck, Darmstaadt, Alemania), MgO (pureza>99%, Merck, Darmstaadt, Alemania) y Ca3(PO4)2 (Carlo Erba, Barcelona, España), se mezclaron para llegar a la composición dada en la tabla 2. A partir de la mezcla en polvo, se conformaron cilindros cerámicos mediante compactación isostática en frío; los cilindros se sinterizaron a una temperatura de 1.200°C, en un horno de marca Hobersal, obteniendo un material compacto.
Para obtener materiales densos, tanto cristalinos como vítreos, se empleó una técnica de procesado láser, LFZ. En esta técnica, un haz láser se focaliza sobre un precursor generando una zona fundida, que se mantiene flotante por tensión superficial. Mediante el desplazamiento de dicha zona fundida a lo largo de la muestra, se obtiene un cilindro solidificado direccionalmente [24]. El proceso de crecimiento se lleva a cabo dentro de una cámara con atmósfera controlada, a partir de precursores cerámicos sinterizados y empleando un láser de CO2 (Blade-600, Electronic Engineering, S.p.A., Florencia, Italia). Los precursores cerámicos se sujetan sobre unos ejes con movimientos independientes de traslación y rotación. Para analizar la influencia de la velocidad de crecimiento en la microestructura del material solidificado, se crecieron muestras a 2velocidades diferentes, 25 y a 300mm/h, obteniendo 2muestras M25 y M300, respectivamente.
La microestructura del material procesado se estudió mediante microscopía electrónica de barrido de emisión de campo (MEB-EC) (Carl Zeiss MERLIN™, Carl Zeiss MicroImaging GmbH, Jena, Alemania). Se cuantificaron los siguientes elementos, Mg, P, Ca, Si y O mediante microanálisis por energía dispersiva de rayos X (EDS) (INCA 350, Oxford Instruments, High Wycombe, Reino Unido).
Se llevó a cabo el estudio de las fases cristalinas presentes en la muestra crecida a 25mm/h por difracción de rayos X (DRX). Los datos se han recogido a temperatura ambiente usando un difractómetro RIGAKU, modelo D-Max/2500 (Rigaku Corporation, Tokyo, Japón) provisto de un ánodo rotante. El difractómetro funciona a 40 kV y 80mA con un ánodo de Cu. Las condiciones de medida fueron: 2 theta de 5˚ a 80˚, step=0,03˚ y t=1 s/step.
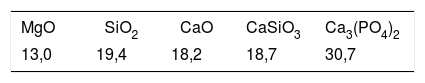
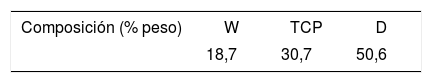
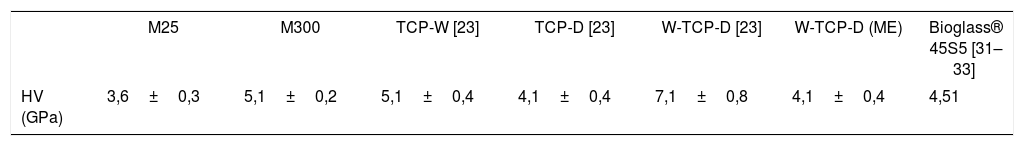
Se determinó la microdureza de estos materiales procesados a 2velocidades diferentes, de este modo podemos determinar la influencia de la microestructura de la muestra en las propiedades mecánicas de la misma. Para ello se realizó un ensayo de dureza Vickers (HV), con un microdurómetro Matsuzawa MXT70 (Matsuzawa Co. Ltd., Kawabe,Akita-shi, Akita Pref, Japón), aplicando una carga de 200 y 300 gf durante 15 s y utilizando un indentador de diamante en forma de pirámide tetragonal se realizaron 10indentaciones, siendo el resultado presentado la media de ellos. Los valores obtenidos se compararon tanto con valores de HV extraídos de la literatura, así como con una muestra vítrea con composición eutéctica del sistema ternario W-TCP-D (ME), mostrada en la tabla 3 y obtenida con la misma técnica que las muestras M25 y M300. Según la literatura, las muestras TCP-D, W-D y W-TCP-D se obtuvieron por fusión durante una hora a temperatura de entre 1.400 y 1.600 ˚C, dando lugar a vidrios transparentes y homogéneos. Sus composiciones se presentan en la tabla 4[23].
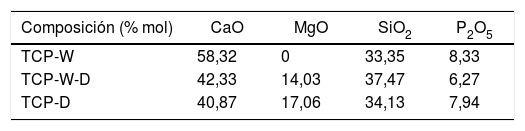
Composición materiales TCP-W, TCP-W-D, TCP-D
| Composición (% mol) | CaO | MgO | SiO2 | P2O5 |
|---|---|---|---|---|
| TCP-W | 58,32 | 0 | 33,35 | 8,33 |
| TCP-W-D | 42,33 | 14,03 | 37,47 | 6,27 |
| TCP-D | 40,87 | 17,06 | 34,13 | 7,94 |
Fuente: Jugdaohsingh [9].
Para el estudio de bioactividad de las muestras procesadas, se empleó el SFA propuesto por Kokubo et al. [25,26] de composición: Na+ 142,0, K+ 5,0, Mg2+ 1,5, Ca2+ 2,5, Cl− 147,8, HCO− 5,0, HPO42− 1,0 y SO42− 0,5mol m–3. Los cilindros solidificados direccionalmente por LFZ se cortaron en discos, en dirección transversal al eje de crecimiento, y tras lavarse con acetona, se colocaron en SFA a 37 ˚C y pH=7,2-7,4, usando HCl para ajustar el pH. Los discos se sacaron del SFA transcurridas 4semanas y se secaron en aire a temperatura ambiente. La preparación metalográfica, para el estudio de la microestructura y el ensayo de dureza, se realizó colocando discos cilíndricos de cada una de las muestras, en un porta muestras de aluminio y llevando a cabo un proceso de desbastado y pulido, mediante discos de carburo de silicio siguiendo la secuencia 120, 240, 400, 800 y 1.200 p, y paños con partículas de diamante de 3, 1, 0,25μm. Se llevó a cabo el análisis de su sección transversal por MEB-EC con 15KeV y se realizó la determinación cuantitativa de calcio (Ca), magnesio (Mg), silicio (Si), fósforo (P), oxígeno (O) mediante EDS, de las mismas.
Resultados y discusiónEstudio de la microestructura de los cilindros solidificados direccionalmenteEn este apartado se exponen los resultados obtenidos del estudio de la influencia de la velocidad de crecimiento en la microestructura de las muestras generadas.
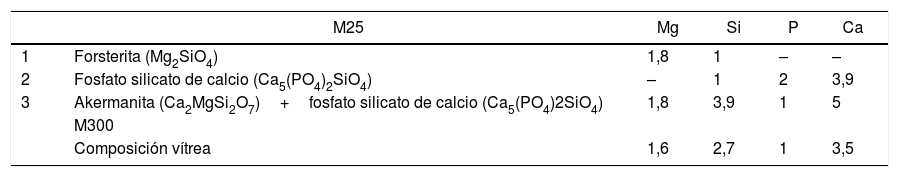
Llevamos a cabo la identificación de fases cristalinas presentes en la muestra crecida a 25mm/h (M25) mediante DRX, los resultados obtenidos se exponen en la figura 1. Según estos resultados, las fases cristalinas que se identifican en la muestra son akermanita (Ca2MgSi2O7) y fosfato silicato de calcio (Ca5(PO4)2SiO4). Para la determinación de fases se ha utilizado la base de datos JCPDS-International Centre for Diffraction Data-2000. En la figura anteriormente mencionada se compara el difractograma experimental (negro) con el difractograma teórico (rojo) resultante de la suma de fases encontradas en la búsqueda.

En la figura 2 se presentan imágenes de MEB de M25, tomadas a diferentes aumentos, 200X (a), 1KX (b) y 8KX (c), y de la muestra procesada a 300mm/h (d) a 200X.
 y 300mm/h (M300). Imágenes de MEB de M25 a 200X (a), 1KX (b) y a 8KX (c) e imagen de MEB de M300 a 200X (d).")
En las imágenes correspondientes a M25 (a, b y c) se aprecia la formación de una estructura formada por 2fases, una más oscura (1) y otra más clara (2), y un constituyente eutéctico (3), señalados en la figura 2 b. La composición de cada una de estas fases, obtenida por EDS, se presenta en la tabla 5. Según los resultados que se presentan en la tabla, y apoyándonos en los resultados de DRX, podemos concluir que nuestro material cristalino está constituido fundamentalmente por un constituyente eutéctico akermanita-fosfato silicato de calcio (3), con pequeñas zonas ricas en forsterita (1) y otras zonas minoritarias en las que encontramos una fase rica en fosfato silicato de calcio (2). Las fases 1 y 2 se encuentran distribuidas homogéneamente a lo largo de la muestra, presentan forma alargada y tienen aproximadamente 6μm de anchura, ambas son fases minoritarias. La fase mayoritaria es la fase 3, el constituyente eutéctico, que se dispone formando colonias relativamente homogéneas en tamaño a lo largo de la muestra, la anchura de esas islas es de aproximadamente 25μm.
Composición de cada una de las fases presentes en la estructura del material procesado a 25mm/h (M25) y 300mm/h (M300)
| M25 | Mg | Si | P | Ca | |
|---|---|---|---|---|---|
| 1 | Forsterita (Mg2SiO4) | 1,8 | 1 | – | – |
| 2 | Fosfato silicato de calcio (Ca5(PO4)2SiO4) | – | 1 | 2 | 3,9 |
| 3 | Akermanita (Ca2MgSi2O7)+fosfato silicato de calcio (Ca5(PO4)2SiO4) | 1,8 | 3,9 | 1 | 5 |
| M300 | |||||
| Composición vítrea | 1,6 | 2,7 | 1 | 3,5 |
La muestra crecida a velocidad mayor (300mm/h, M300) presenta una estructura vítrea, tal y como se observa en la figura 2 (d), donde se aprecia la formación de una única fase. La composición atómica, obtenida por EDS, de la muestra procesada a 300mm/h es la que se presenta en la tabla 5.
A mayor velocidad de enfriamiento se obtiene una fase amorfa, mientras que a menor velocidad de enfriamiento se forman fases cristalinas. En el proceso de solidificación, en este último caso, se forman dendritas de forsterita y fosfato silicato de calcio, dando lugar a un líquido con composición eutéctica. Este solidifica en 2fases de akermanita y fosfato silicato, las cuales crecen de forma cooperativa con una estructura laminar.
Estudio de microdureza VickersEn este apartado vamos a exponer los resultados del estudio de microdureza a temperatura ambiente, para cada una de las muestras procesadas, M25 (procesada a 25mm/h) y M300 (procesada a 300mm/h). Los resultados obtenidos se comparan con los de otros materiales vítreos estudiados para el mismo fin (W-TCP, TCP-D, TCP-W-D y Bioglass® 45S5; marca registrada de la Universidad de Florida).
En la tabla 6 se presentan estos valores. En ella podemos observar que la dureza obtenida para el material vítreo (M300) es mayor que la del TCP-D, el Bioglass®, y el ternario TCP-W-D (ME) y presenta una dureza semejante al TCP-W. Con todo esto podemos concluir que la dureza de nuestro material es apta para emplearse como implante óseo. La dureza del material cristalino (M25) es menor que la del vítreo y menor que las del resto de los vidrios que se presentan en la tabla 6; esto es debido a que mayor dureza se corresponde con una mayor homogeneidad de la muestra. La mayor homogeneidad se presenta en el material vítreo, ya que el material cristalino presenta un mayor número de defectos, poros, grietas y fases.
Como hemos visto en el apartado anterior somos capaces de controlar la microestructura de la muestra al modificar la velocidad de crecimiento, por tanto, podremos adecuar la dureza de la misma, ya que según se presenta en la tabla 6 la dureza obtenida para muestras crecidas a mayor velocidad (M300) es mayor que para las muestras crecidas a menor velocidad (M25).
Estudio de bioactividad de las muestras procesadasPara estudiar la bioactividad de los cilindros solidificados direccionalmente, sumergimos en SFA varios discos de ambas muestras y tras 4semanas analizamos por MEB los posibles efectos del SFA en ellas.
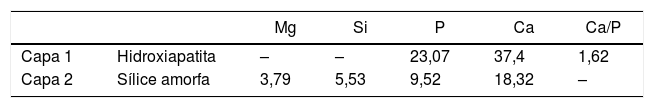
Por un lado, la figura 3 corresponde a una imagen de MEB de un corte transversal de una muestra cristalina obtenida por crecimiento lento (25mm/h), tras el ensayo de bioactividad. En esta imagen se puede apreciar la formación de 2regiones diferentes del material de partida. La región más externa (región 1) está formada por 2capas (capas 1 y 2), cuya composición se obtuvo por EDS y se presenta en la tabla 7. Podemos observar que mientras en la capa más externa de dicha región (capa 1) no encontramos Si, en la capa 2 hay un alto porcentaje de este elemento. La composición de la capa 1 corresponde a HA (con una relación Ca/P=1,62), la composición de la capa 2 sería compatible con la formación de una capa de sílice amorfa. Tal y como se observa en la imagen, la segunda región, presenta una cierta porosidad interconectada hasta un espesor de aproximadamente 37μm, generada por la disolución parcial de akermanita en contacto con SFA, dejando huecos de tamaño submicrométrico.
 tras el ensayo de bioactividad. Región 1, incluyendo capa 1 (HA), capa 2 (sílice amorfa) y región 2.")
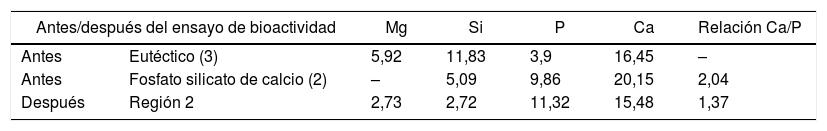
En la tabla 8 se puede observar la comparación de composición de esta segunda región con la zona correspondiente antes de llevar a cabo el estudio en SFA. En esta tabla se observa la disminución de la concentración de magnesio, solo presente en la akermanita. Por otra parte, la disminución de la concentración de Si y la disminución de la relación Ca/P hace pensar que se produce una trasformación de fosfato silicato de calcio a HA al estar en contacto con sistemas fisiológicos. Dicha transformación no se ha observado en las fases primarias de fosfato silicato de calcio debido a que la superficie específica es menor que en las lamelas del constituyente eutéctico.
Comparación de la composición de la muestra M25 antes y después del ensayo de bioactividad
| Antes/después del ensayo de bioactividad | Mg | Si | P | Ca | Relación Ca/P | |
|---|---|---|---|---|---|---|
| Antes | Eutéctico (3) | 5,92 | 11,83 | 3,9 | 16,45 | – |
| Antes | Fosfato silicato de calcio (2) | – | 5,09 | 9,86 | 20,15 | 2,04 |
| Después | Región 2 | 2,73 | 2,72 | 11,32 | 15,48 | 1,37 |
Tal y como propone Kokubo et al. [26], la formación de una capa de HA podría generar una respuesta biológica en la interfase entre la HA y el tejido óseo, estableciendo una unión biológica entre un implante y el hueso favoreciendo, de este modo, la regeneración del tejido dañado.
Además, al generarse cierta porosidad en la muestra cristalina tras el ensayo de bioactividad, la dureza de nuestro material en contacto con sistemas fisiológicos se hará menor aproximándose a la del hueso (0,42GPa) [27].
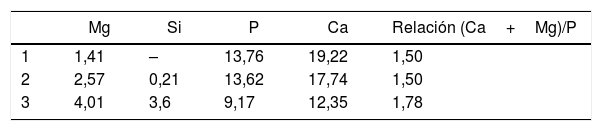
Por otro lado, en la figura 4 podemos observar el efecto del SFA en la muestra vítrea obtenida por procesamiento láser a 300mm/h. En esta imagen, de igual modo que en el caso de la muestra crecida a menor velocidad, se puede observar la generación de varias capas tras el ensayo de bioactividad. La composición de estas capas se muestra en la tabla 9. Observando los resultados que se presentan, se puede concluir que realmente se han producido cambios en la superficie de la muestra al estar en contacto son SFA. Estos cambios consisten en la formación de sucesivas capas (capas 1, 2 y 3), cuyo contenido en fósforo aumenta conforme nos acercamos a la capa más externa (capa 1) y el contenido en silicio y magnesio disminuye; este hecho evidencia que estos elementos experimentan una disolución en el SFA. La relación (Ca+Mg)/P obtenida, para las capas 1 y 2 corresponde a HA deficiente en calcio (CDHA). En la capa 3, la composición cambia con respecto a la del vidrio original, durante esta transformación se produce una disminución del contenido en Mg, Si, Ca, aproximándose a la composición de la HA.
 tras el ensayo de bioactividad. Capas 1, 2 y 3 (CDHA) con contenido creciente en Si.")
El espesor de la zona afectada es mayor para el caso del material vítreo (60μm), si consideramos todas las capas formadas, que para el material cristalino (52μm), teniendo en cuenta tanto la región 1 como la región 2, para el mismo tiempo de inmersión en SFA.
La formación de la capa de HA, tanto en el material vítreo como en el cristalino (M300, M25), se puede explicar según en mecanismo propuesto por Hench et al. [8] para el caso de materiales vítreos. Dicho mecanismo puede resumirse en los siguientes pasos: 1) intercambio rápido de iones alcalino y alcalinotérreos con H+ de la solución; 2) pérdida de sílice en forma de Si(OH)4 a la solución; 3) condensación y repolimerización de una capa rica en SiO2 en la superficie pobre en cationes alcalinos y alcalinotérreos; 4) migración de grupos Ca2+ y PO43− a la superficie, formando una capa de CaO-P2O5 sobre la capa rica en SiO2, seguida por la incorporación de calcio y fósforo soluble de la solución, y 5) cristalización de la capa amorfa de CaO-P2O5 por la incorporación de iones OH− de la solución para formar una capa de HA [28–30].
Como podemos observar en las figuras 3 y 4, la reactividad del material vítreo (M300) es mayor que la del material cristalino (M25), ya que para el mismo tiempo de inmersión en SFA la zona afectada alcanza una mayor profundidad.
ConclusionesAl final del trabajo se pueden establecer las siguientes conclusiones:
- –
A partir de una composición desviada de la composición eutéctica y modificando la velocidad de crecimiento se ha podido ejercer un control sobre la microestructura de las muestras y sobre sus propiedades mecánicas y de bioactividad.
- –
Cuando las muestras se crecen a velocidad lenta (25mm/h) poseen estructura cristalina y crecidas a mayor velocidad (300mm/h) tienen estructura vítrea.
- –
Las muestras obtenidas son bioactivas, ya que en contacto con SFA son capaces de generar una capa de HA en la superficie del material cuya función es promover la unión del implante al hueso y la regeneración del tejido dañado.
- –
Tras el ensayo de bioactividad, la muestra vítrea se mantiene densa y genera una capa de HA gruesa, mientras que la muestra cristalina genera una porosidad por disolución parcial de una de las fases y la formación de una capa de HA.

