













Introducción: La calcinosis tumoral es una enfermedad rara, cuya prevalencia es difícil de determinar debido a lo aislado de los casos. Se caracteriza por depósitos periarticulares, únicos o múltiples, de cristales de calcio y fosfatos. Esto es debido a una falla en la inhibición de la recaptura de fosfatos a nivel de los túbulos proximales, lo que causa hiperfosfatemia. No está descrito un tratamiento óptimo y el reducido número de casos dificulta la evaluación de los resultados.
Caso clínico: Se presenta el caso de una paciente de sexo femenino de 10 años de edad, con un cuadro clínico caracterizado por aumento de volumen en ambos codos y en el dedo índice de la mano derecha. Se realizó diagnóstico de calcinosis tumoral hiperfosfatémica. Se dio tratamiento quirúrgico y médico, sin presentar recidiva en los primeros seis meses de seguimiento.
Conclusiones: La calcinosis tumoral es una entidad rara que debe sospecharse al tener un paciente con calcificaciones periarticulares únicas o múltiples. El tratamiento médico debe orientarse a restablecer el equilibrio calcio-fósforo. El tratamiento quirúrgico se utiliza por razones estéticas y funcionales.
Background: Tumoral calcinosis is a rare disease whose prevalence is hard to determine due to the scarcity of the reported cases. This disease is distinguished by single or multiple periarticular deposits of calcium and phosphate due to a failure in the phosphate recapture inhibition at the proximal renal tubules, resulting in hyperphosphatemia. There is no optimal treatment described because there are a reduced number of cases, which makes it difficult to assess the results.
Case report: We present the case of a 10-year-old female with a growing mass in both elbows and index finger of the right hand. Diagnosis was made of hyperphosphatemic tumoral calcinosis. Surgical and medical treatment were given, with no relapse during a 6-month follow-up.
Conclusions: Tumoral calcinosis is a rare entity that should be considered when a patient has a single or multiple periarticular calcifications. Medical treatment is oriented to restoring the calcium-phosphate balance. Surgical treatment should be offered for aesthetic and functional reasons.
1. Introduction
Tumor calcinosis is a rare disease that was first described in 1943 by Inclan et al.,1 although there are reports of similar cases reported in 1889 by Duret,2 and in 1953 by Teutschlaender who called it lipocalcinogranulomatosis.3 Its prevalence is difficult to determine because cases are isolated. A greater incidence has been observed in Afro-American patients, in the first and second decade of life, and without gender predominance.
Under normal conditions, serum phosphate levels depend on three factors: intestinal absorption, skeletal storage and renal reabsorption. These processes in turn are regulated by three endocrine factors: fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH) and calcitriol (1,25(OH)2D3).4
Phosphate absorption in the intestine is mediated by 1,25-(OH)2D3. Serum phosphate is taken up by the cells by means of type II and III co-transporters of sodium-phosphate to carry out functions such as DNA synthesis and lipid membrane oxidation among others.5 Another portion of serum phosphate is stored in the skeleton in the form of hydroxyapatite crystals. Phosphate reabsorption in the kidneys is mediated by PTH and FGF23 and is carried out in the proximal tubules by means of type II and III sodium/phosphate co-transporter. Type II sodium/phosphate co-transporter is responsible for 70-80% of phosphate reabsorption.4,5
FGF23 is an endocrine factor with hormonal activity that reduces serum phosphate concentration, decreasing its intestinal absorption and reabsorption in the proximal tubules. FGF23 is produced in the osteocytes, osteoblasts and osteoprogenitor cells and is regulated by PHEX (phosphate regulating endopeptidase homolog, X-linked) and DMP1 (dentin matrix protein 1).6 Before FGF23 is secreted in the circulation it passes through a stage of O-glycosylation mediated by the GALNT3 protein (polypeptide N-acetylgalactosamine transferase 3) in order to be transformed into its active form. FGF23 unites in the proximal tubules together with Klotho alpha (α-KL) to the FGFR1c receptor (fibroblast growth factor receptor 1c). Activation of this receptor decreases expression of type IIa and IIc sodium/phosphate co-transporters, inhibiting phosphate reabsorption. FGF23 also reduces the synthesis of 1,25(OH)2D3, decreasing intestinal absorption of calcium and phosphate.5
Hyperphosphatemic familial tumoral calcinosis is an autosomal recessive disease characterized by peri articular deposits of calcium and phosphate crystals in soft tissues due to an increase in the serum phosphate concentration.7 Hyperphosphatemia is due to phosphate reuptake not being inhibited at the level of the proximal tubules by means of FGF23.5 Three different mechanisms for the poor functioning of FGF23 have been described. In type I there is a homozygous mutation in the gene that codifies the GALNT3 protein, for which glycosylation of FGF23 is not carried out. Active serum levels of FGF23 are found to be decreased, whereas nonglycosylated levels of FGF23 are elevated. In type 2 a homozygous mutation in FGF23 results in an elevated nonfunctional serum level of FGF23. In type 3 there is a homozygous mutation of the gene that codifies α-KL, the enzyme that, together with FGF23, activates the FGFR1c receptor for which, even though there is intact circulating FGF23, its signal cannot be transmitted through the receptor.5 Together these mechanisms allow an increase in the reabsorption of renal phosphate and, as a result, hyperphosphatemia. Among the clinical findings that may be observed due to hyperphosphatemia are formation of calcium deposits, periarticular phosphate and, in soft tissues, calcifications in the dental pulp and in the eyelids, vascular calcifications, nephrocalcinosis and bone mineral density.8 Nonpainful periarticular tumors progressively increase in size (over months or years), generally on the extensor side of the joints without causing limitations in movement. The joints most affected are the hip, shoulder and elbows, although any joint can be affected. The skin that is found underlying the lesions may present fistula tracts, with intermittent drainage of caseous material that spontaneously ceases.9
Radiographic images typically demonstrate amorphous, cystic, multilobulated calcifications.10
Laboratory exams do not show any alterations in serum calcium levels. However, serum phosphorus may be normal or increased with increased renal absorption of phosphorus. Parathyroid hormone levels are found within normal limits. Renal function is preserved.
Anatomopathological findings show lobulated, yellowish-white calcified masses. On microscopy, calcifications surrounded by chronic inflammatory reaction due to foreign body with giant cells are present.10
Due to the low incidence of this condition, treatment guidelines have not been established. However, there have been cases reported where medical treatment has been successful.11-13 On the one hand, decrease of intestinal absorption of phosphate is sought by restricting the diet (to 400 mg/day plus aluminum hydroxide and/ or sevelamer).4 Use of acetazolamide is recommended for inhibiting renal absorption of phosphate.4,5 When the lesions cause pain, deformity or limitation of movement, surgical resection of the deposits is recommended.14-18 However, the lesions tend to recur if the metabolic derangement remains.4 Therefore, resection together with medical treatment is recommended.19,20
2. Clinical case
The case of a 10-year-old female is presented. The patient is from Matamoros, Tamaulipas, Mexico. She was previously healthy without any significant past medical history and without history of endogamy in the family or family members with lesions similar to the ones she presented.
She presented for consultation due to a 1-year increase in volume on the extensor side of the right elbow, increased volume on the extensor side of left elbow for 6 months, discharge of caseous material intermittently from the left elbow for 3 months, and increased volume in the index finger of the right hand 2 months prior. During this time the patient had no fever, weight loss, weakness, myalgias, pain or limitation of movement of the affected joints. She received two schemes of antibiotics without improvement. Written informed consent was obtained from the patient's mother for this case report and accompanying images.
Physical examination found both elbows with an increase in volume in their extensor face with the lesions demonstrating soft consistency. The range of motion was found without limitation in flexion or extension and no pain on movement (Figs. 1 and 2). There were multiple lesions on the left elbow, which corresponded to exit tracts of caseous material. There were no reports of infection and no discharge of caseous material at that time (Figs. 3 and 4). The mass of the right index finger demonstrated yellowish-white, soft lesions that were painful to palpation. There were no lesions, tumor or dermatosis found in the remainder of the joints. Data of calcinosis in the teeth and eyes was intentionally sought without significant findings.
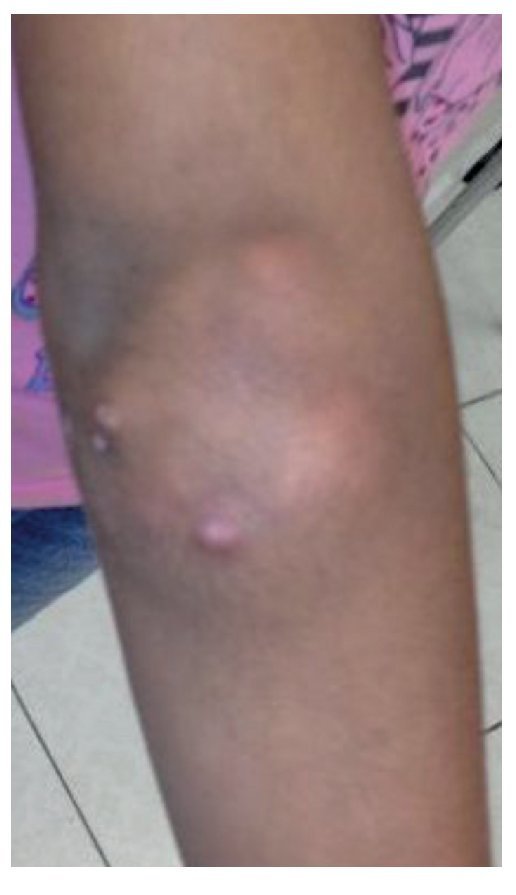
Figure 1 Frontal view of the right elbow.
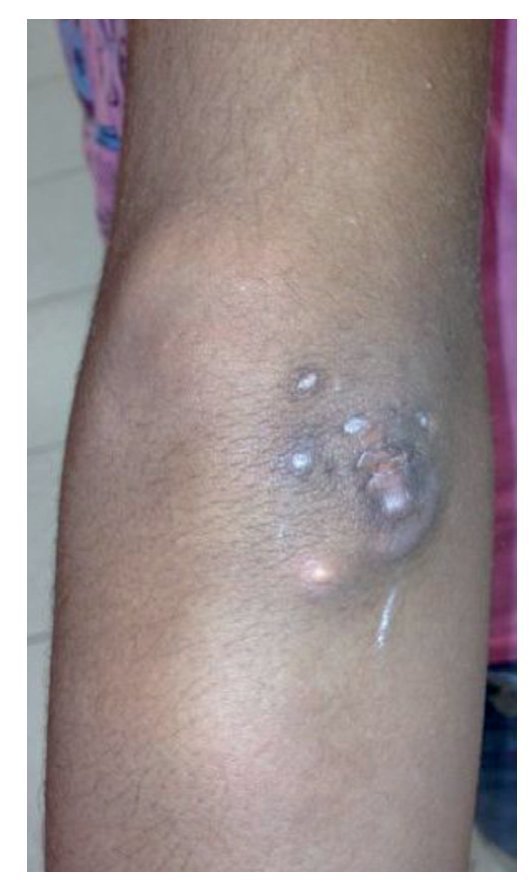
Figure 2 Lateral view of the right elbow.
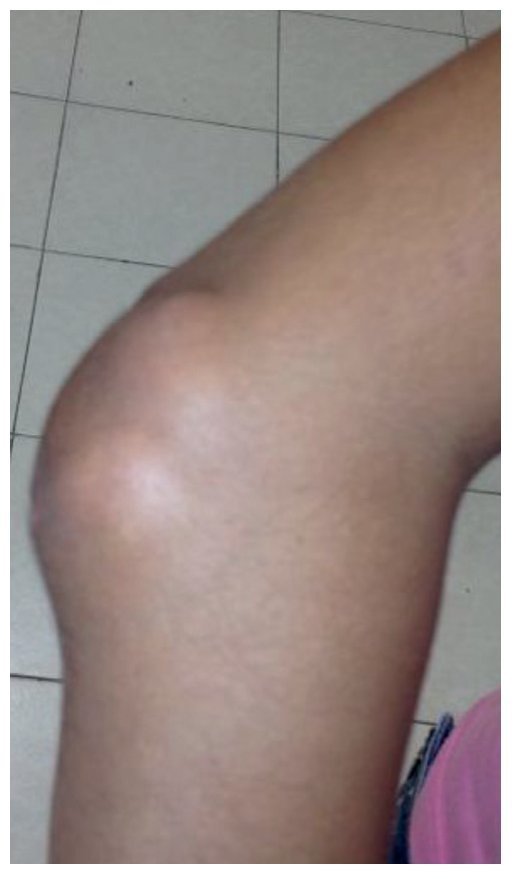
Figure 3 Frontal view of left elbow.
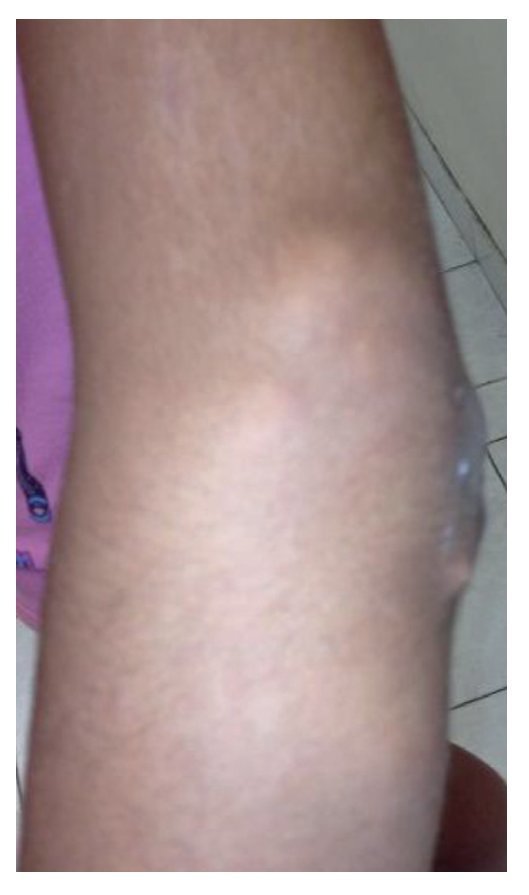
Figure 4 Lateral view of left elbow.
X-rays were taken of the elbows (Figures 5 and 6) and of the right hand (Figure 7) in which conglomerates of multiple, round homogeneous opacities separated by radiolucent lines that did not affect or depend upon the adjacent bony structures were seen. X-rays of the chest and abdomen were requested as well as echocardiogram, renal ultrasound and plain CT of the skull in search of calcifications. All studies were reported to be without significant findings.
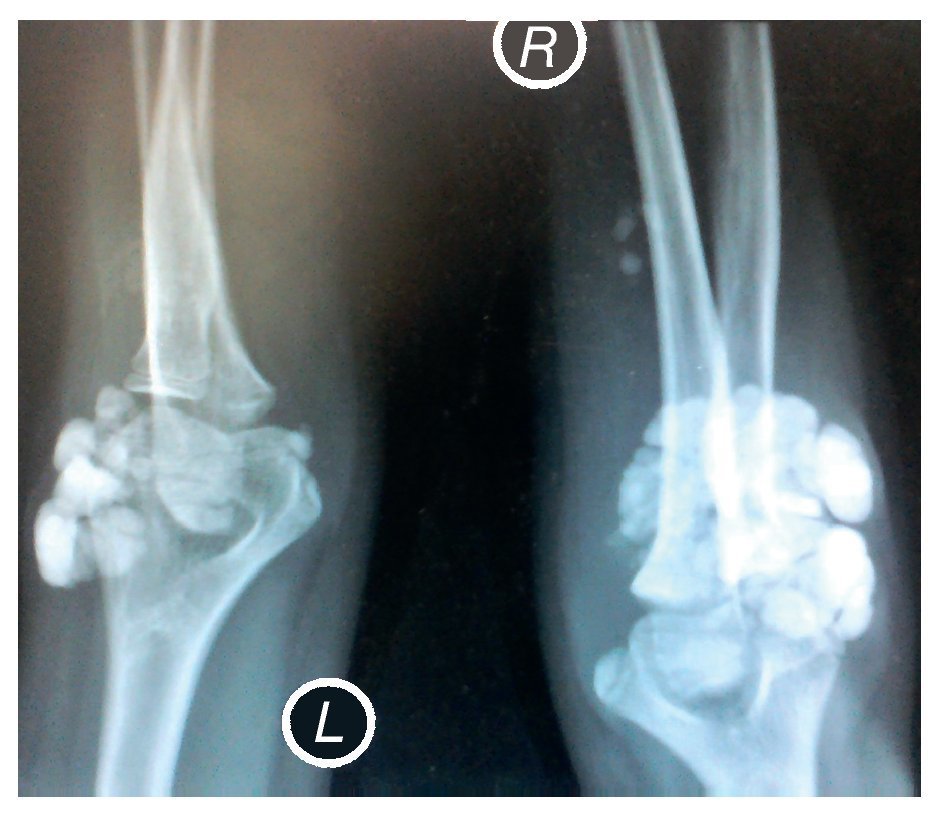
Figure 5 Anteroposterior (AP) x-ray of both elbows (L: left; R: right).
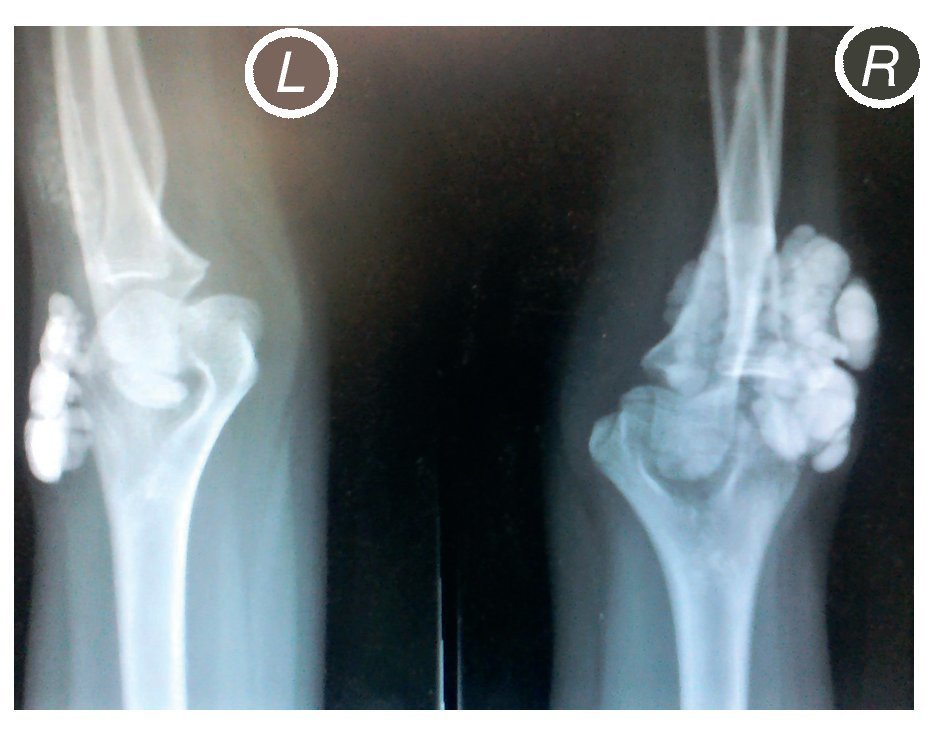
Figure 6 Lateral x-ray of both elbows (L: left; R: right).
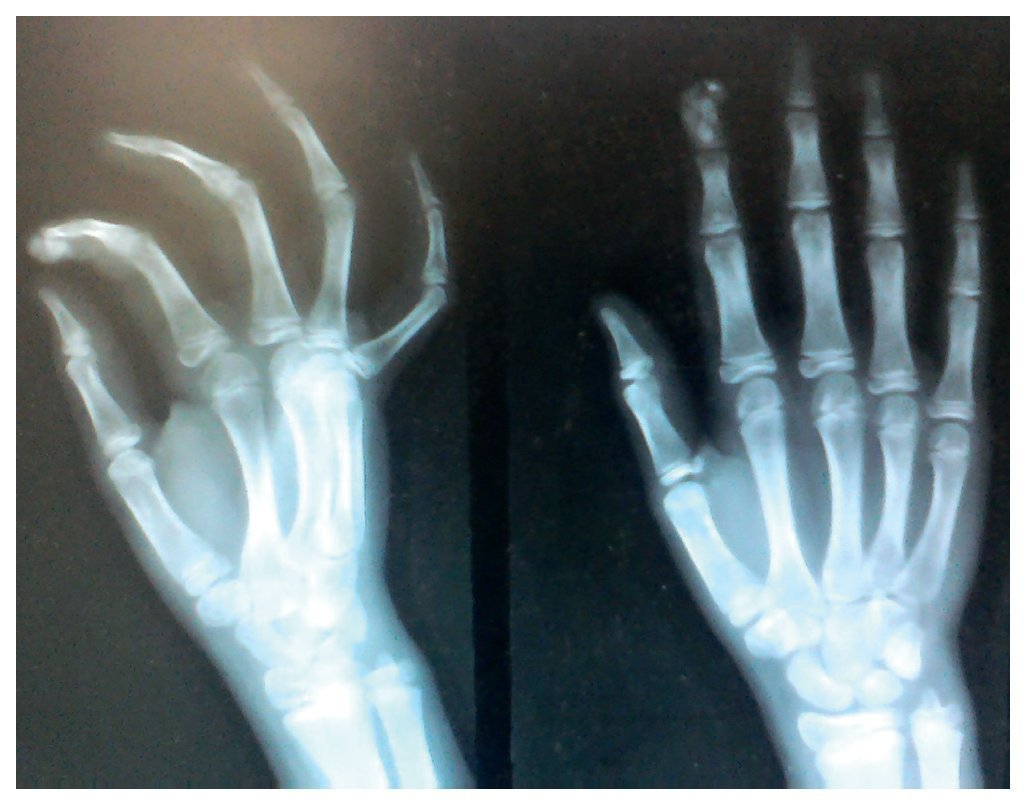
Figure 7 AP and lateral x-ray of the right hand.
Laboratory test results showed normal serum creatinine (0.28 mg/dl), normal serum sodium (139 mmol/l), normal potassium (4.1 mmol/l), normal calcium level (9.8 mg/dl), hyperphosphatemia (5.9 mg/dl), normal parathyroid hormone level (27.5 pg/ml), decreased 24-h urine phosphorus (307 mg/dl), maximum threshold of tubular phosphate reabsorption 5.72 mg/dl, increased tubular phosphorus reabsorption (97.1%) and glomerular filtration rate corrected for body surface of 293.79 ml/min/m2. Diagnosis of hyperphosphatemic tumoral calcinosis was made.
Complete surgical resection of the elbow calcifications was carried out and the specimens were sent for pathological examination. On macroscopic examination, grayish-white lobulated fragments of lax consistency were reported (Fig. 8). The cut surface of the section was heterogeneous and multiple whitish nodules of caseous appearance and friable consistency on a fibrous background were seen (Fig. 9). Histopathological examination with hematoxylin and eosin reported large masses of calcification deposited in the connective stroma surrounded by reaction to foreign body and fibroblastic activity (Fig. 10). There were no osteocytes or osteoblasts seen (Fig. 11).
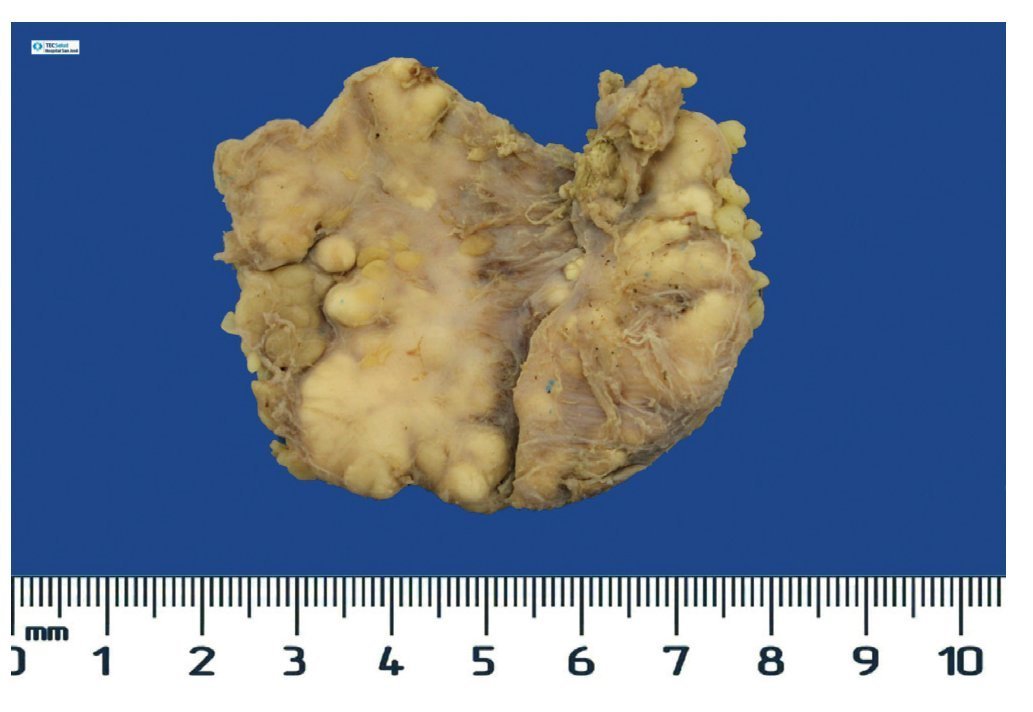
Figure 8 Irregular soft tissue tumor of the right elbow (24.2 g and 6.0 × 4.5 × 2.0 cm).
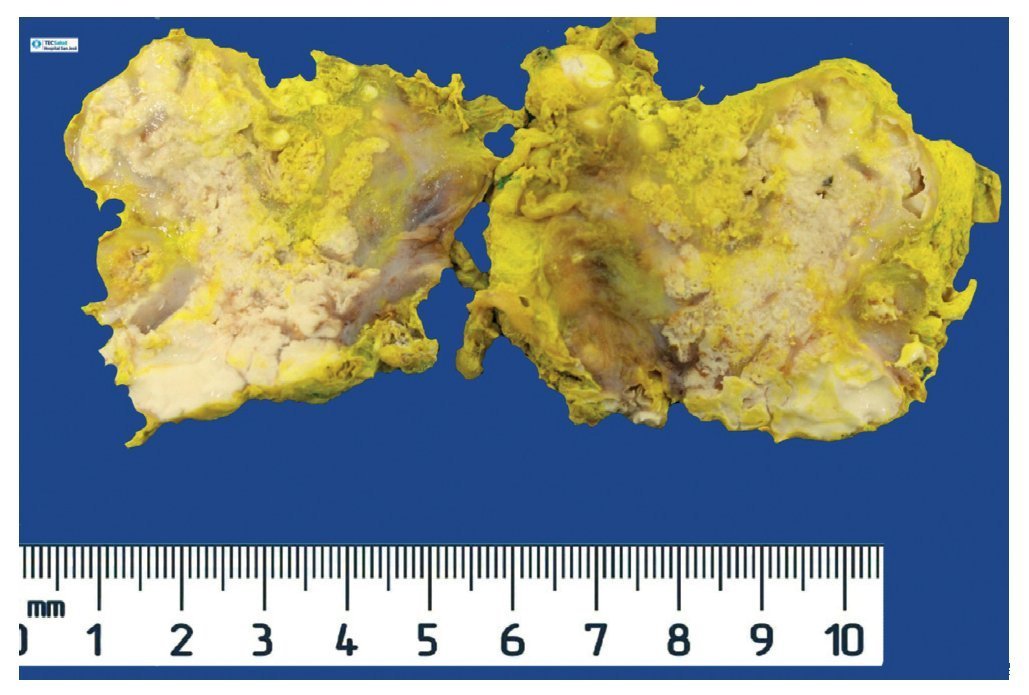
Figure 9 Tumor is multilobulated and heterogeneous on cut with friable consistency.
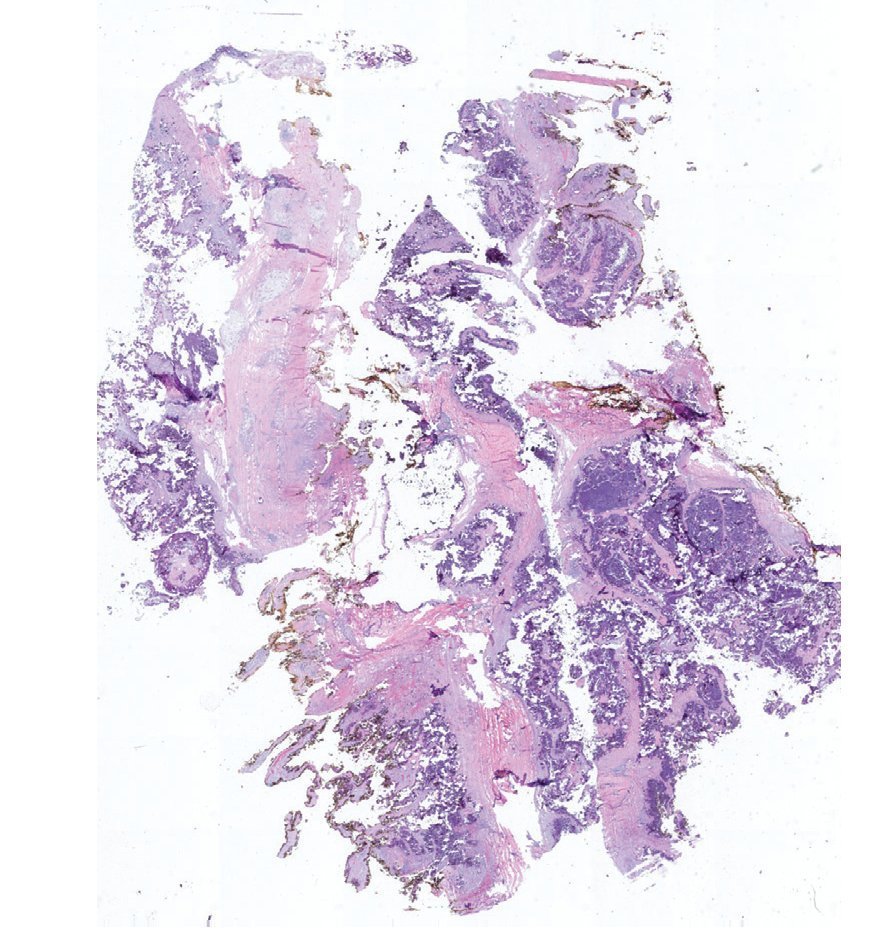
Figure 10 Representative cut of the lesion demonstrating abundant dystrophic calcification surrounded by fibrous tissue. Circumferential resection border in yellow (1.25× HE).
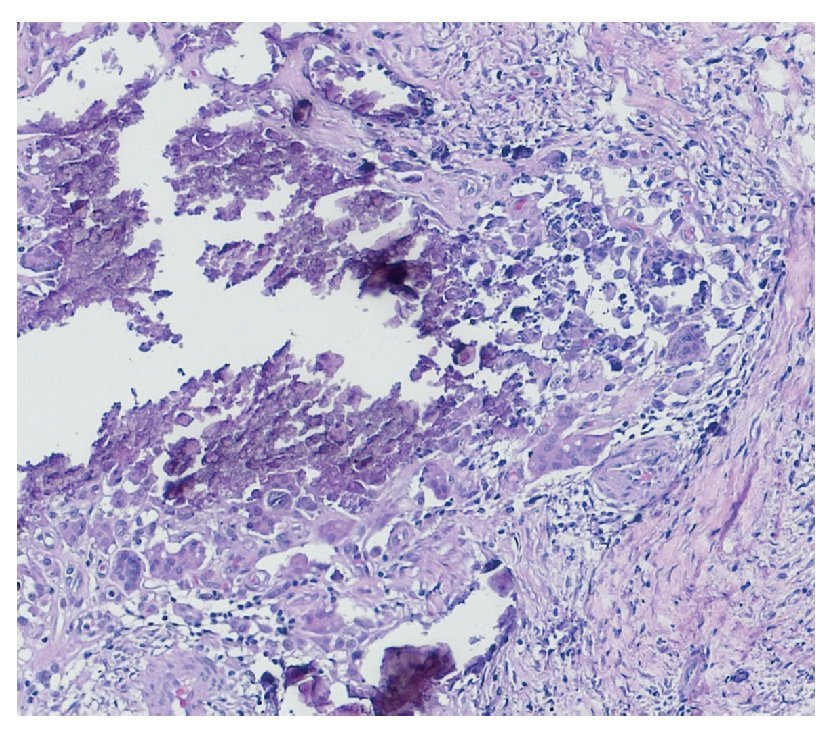
Figure 11 Dystrophic calcification surrounded by multi-nucleated giant cells of foreign body type and chronic inflammation (40× HE).
Subsequent to the resection of the elbow calcifications, the patient was discharged with treatment with 10 mL of hydroxide aluminum and magnesium four times a day prior to meals and a diet low in phosphorus. Telephone follow-up at 6 months after beginning of treatment was done. It was reported that there was no recurrence of the elbow calcifications and that the calcification on the index finger of the right hand had greatly decreased until it was almost imperceptible. The same management was continued.
Discussion
The presence of periarticular calcifications, alone or multiple, is a rare finding in pediatrics. Tumoral calcinosis should be suspected in the differential diagnosis together with other diseases that can share its radiological charac teristics, distribution, size and morphology, mainly connective tissue diseases (dermatomyositis, scleroderma, lupus, CREST syndrome), neoplastic diseases (synovial sarcoma, osteosarcoma) and chronic renal insufficiency.
However, the clinical history of tumoral calcinosis is very remarkable with nonpainful periarticular tumors that progressively increase in size over months or years, generally appearing on the extensor side of the joints without limitation of movement. Radiological findings showed lobulated periarticular calcifications separated by radiolucent lines on plain films and laboratory results demonstrated normocalcemia and hyperphosphatemia or normophosphatemia, with normal renal and parathyroid functions.
Scleroderma is considered among the differential diagnoses of calcinosis. Scleroderma is an autoimmune disease of the connective tissues that involves damage to the skin, blood vessels, muscles and internal organs. In pediatric-age patients, the main characteristics are hardened skin and appearance before age 16 years. It is divided into two categories: localized scleroderma where there is involvement of the skin but not of internal organs or vascular system, and systemic scleroderma, which does involve internal organs, particularly the esophagus, GI tract, heart, lungs and kidneys.21 For the diagnosis of systemic scleroderma certain preliminary criteria are used.21
Among the major criteria are the following:
• Tissue sclerosis or induration
• Sclerodactyly
• Raynaud's phenomenon
Among the minor criteria are the following:
• Vascular changes (changes in nails, capillaries and digital ulcers)
• Gastrointestinal involvement (dysphagia, gastroesophageal reflux)
• Renal involvement (renal crisis, new-onset hypertension)
• Cardiac involvement (arrhythmias, cardiac insufficiency)
• Pulmonary involvement (pulmonary fibrosis, pulmonary hypertension)
• Musculoskeletal involvement (joint friction, arthritis, myositis, subcutaneous calcifications)
• Neurological involvement (neuropathy, carpal tunnel syndrome)
• Serological involvement (antinuclear antibodies, antibodies specific for systemic sclerosis)
The patient in this case presented only one minor criterion for systemic scleroderma, for which the probability of this diagnosis was low.
In localized scleroderma, the lesions are commonly confined to the dermis, with small deep involvement. Isolated cases have been published where internal organs are involved in patients with localized scleroderma; however, skin lesions predominate in these cases.21
Diagnosis is made based on the clinical picture and skin or subcutaneous tissue biopsies where the lesion is found. This diagnosis was able to be ruled out because the clinical picture did not follow the pattern for scleroderma, specifically according to the dermatological signs that are required for diagnosis.21
Another diagnostic differential that should be ruled out is dermatomyositis. Juvenile dermatomyositis is the most common juvenile idiopathic inflammatory myopathy. It is an autoimmune disease characterized by a systemic vasculopathy with myositis, progressive muscle weakness and dermatological findings. It is not known with certainty what the etiology of this disease is, but it is believed that it is a combination of genetic predisposition and environmental factors.22
Diagnosis is made using the following criteria:
• Muscular involvement: progressive and symmetric muscular weakness. There may be dysphagia and respiratory involvement.
• Muscle biopsy: type I and II necrosis fibers, phagocytosis, regeneration with basophilia, sarcolemnic nuclei, large vesicles, prominent nucleolus, atrophy with perifascicular distribution, fibers of various sizes, commonly perivascular inflammatory exudates
• Elevation in muscular enzymes: CPK, aldolase, AST, LDH
• Electromyogram: short and small polyphasic motor units, fibrillations, acute positive waves, repetitive discharges of high frequency
• Skin changes: rash in heliotrope with periorbital edema, Gottron sign (erythematous and squamous dermatitis on the dorsum of the hands), involvement of the knees, elbows, medial malleolus, face, neck and proximal thorax.
• Diagnosis is definitive when three or four criteria are met, in addition to the dermatological findings. If there are two criteria present, plus dermatological findings, it is said that there is a probable diagnosis.22
Dystrophic calcinosis is a characteristic complication of juvenile dermatomyositis reported in 30-70% of the patients with the diagnosis already established. Calcifications are mainly found in the elbows and knees and can cause local pain, joint contracture and ulcers of the overlying skin. This complication is due to the damaged muscles secreting calcium to the matrix vesicles and promoting mineralization with accumulation of hydroxyapatite.22 Although this patient had lesions similar to the lesions found in dystrophic calcinosis, she did not fulfill the criteria for the diagnosis of juvenile dermatomyositis.
Systemic lupus erythematosus (SLE), an autoimmune disease of the connective tissue that could affect any part of the body, should also be ruled out. The most common systemic symptoms in pediatric patients are fever, hair loss, fatigue, weight loss, lymphadenopathy and hepatosplenomegaly.23 The organs most affected in children are the skin (malar rash, photosensitivity, vascular lesions, palmar and plantar erythema, Raynaud's phenomena and annular erythema), musculoskeletal (arthritis, arthralgias, tenosynovitis) and renal system.23
The articular lesions related with SLE are characterized by a mild to moderate joint effusion, with decrease in range of motion and that increases pain in the mornings.23 Because the patient did not fit this clinical picture, this pathology was ruled out.
Once the diagnosis of tumoral calcinosis is made, ideal treatment should be sought. It should be taken into account that, currently, the most accepted theory is that the disease is caused by a dysfunction in phosphate secretion at the level of the proximal renal tubules, which causes secondary hyperphosphatemia and massive calcifications in the periarticular regions. Treatment should be oriented in re-establishing calcium-phosphorus metabolism, and surgical treatment should be carried out for aesthetic and functional reasons. Even after a decrease in the calcifications with medical treatment or with surgical resection, there is a risk of recurrence, especially if complete resection was not done and in patients with hyperphosphatemia. For this reason, it is important to provide follow-up to those patients in search of recurrence of the existing calcifications or appearance of new ones. It is also important to follow-up these patients as the cardiovascular prognosis is poor due to accelerated vascular calcification.
In the case presented, surgical treatment of the elbows for aesthetic reasons was decided upon and complete resection of calcifications was performed. Medical treat ment was begun with a diet low in phosphorus and alumi num hydroxide and magnesium to decrease intestinal absorp tion of phosphates. With this, the calcification of the index finger of the right hand decreased; therefore, surgical resection of this calcification was not done.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 7 November 2013;
accepted 2 June 2014
* Corresponding author.
E-mail:dr.abegalindo@gmail.com (A. Galindo Gómez).