









Aniridia: Update in Diagnosis, Complications and Management
More infoAniridia is a panocular disease characterized by iris hypoplasia, accompanied by other ocular manifestations, with a high clinical variability and overlapping with different abnormalities of the anterior and posterior segment. This review focuses on the genetic features of this autosomal dominant pathology, which is caused by the haploinsufficiency of the PAX6 gene. Mutations causing premature stop codons are the most frequent among the wider mutational spectrum of PAX6, with more than 600 different mutations identified so far. Recent advances in next-generation sequencing (NGS) have increased the diagnostic yield in aniridia and contributed to elucidate new etiopathogenic mechanisms leading to PAX6 haploinsufficiency. Here, we also update good practices and recommendations to improve genetic testing and clinical management of aniridia using more cost-effective NGS analysis. Those new approaches also allow studying simultaneously both structural variants and point-mutations in PAX6 as well as other genes for differential diagnosis, simultaneously. Some patients with atypical phenotypes might present mutations in FOXC1 and PITX2, both genes causing a wide spectrum of anterior segment dysgenesis, or in ITPR1, which is responsible for a distinctive form of circumpupillary iris aplasia present in Gillespie syndrome, or other mutations in minor genes. Since aniridia can also associate extraocular anomalies, as it occurs in carriers of PAX6 and WT1 microdeletions leading to WAGR syndrome, genetic studies are crucial to assure a correct diagnosis and clinical management, besides allowing prenatal and preimplantational genetic testing in families.
La aniridia es una enfermedad panocular caracterizada por hipoplasia iridiana, habitualmente acompañada de otras manifestaciones oculares, con gran variabilidad y solapamiento clínico con otras anomalías del segmento anterior y posterior. Esta revisión se centra en las características genéticas de esta enfermedad autosómica dominante causada por mutaciones en el gen PAX6. El espectro mutacional es muy amplio, con más de 700 mutaciones descritas hasta la fecha, siendo las mutaciones que causan un codón de parada prematuro, las más frecuentes. Los recientes avances en las técnicas de secuenciación masiva (NGS) han aumentado el rendimiento diagnóstico en la aniridia, y han contribuido a dilucidar nuevos mecanismos que conducen a la haploinsuficiencia de PAX6. En esta revisión, actualizamos los protocolos de diagnóstico genético incluyendo estas nuevas técnicas de NGS para realizar estudios con mayor coste-efectividad y rapidez. Su uso abre la posibilidad de estudiar simultáneamente las variaciones puntuales y estructurales tanto en PAX6 como en otros genes de diagnóstico diferencial con otras enfermedades solapantes. Algunos pacientes con fenotipos atípicos pueden presentar mutaciones en FOXC1 y PITX2, genes asociados a un amplio espectro de disgenesias del segmento anterior, o en ITPR1, responsable de una forma distintiva de aplasia circumpapilar del iris presente en el síndrome de Gillespie, entre otros. Dado que la aniridia se puede presentar asociada a formas sindrómicas, como el síndrome de WAGR causado por deleciones contiguas de PAX6 y WT1, los estudios genéticos son cruciales para realizar un correcto diagnóstico y manejo clínico de los pacientes, además de permitir diagnósticos prenatales y preimplantacionales en las familias.
Congenital aniridia (MIM #106210) is a panocular disease characterized by abnormal development of the iris and fovea. In general, aniridia presents a high clinical variability with different expressions in the anterior and posterior segment of the eye.1 Patients may exhibit photophobia, nystagmus, strabismus and a visual deficit that may progress to legal blindness. Although it usually presents in isolation, aniridia occurs in one third of patients in a syndromic context, with WAGR syndrome (MIM #194072) being the most frequent form.1
Aniridia is a monogenic disease with an autosomal dominant inheritance pattern that is mostly caused by heterozygous mutations in the PAX6 gene, a key gene in ocular morphogenesis. Molecular studies in aniridia are an essential diagnostic tool for clinical confirmation, patient management and family counselling.2 In recent years, technological advances in the field of genomic medicine have contributed to the description of new molecular mechanisms in the etiopathogenesis of aniridia involving variants in non-coding regions and the demonstration of the existence of mosaicism in some families.3,4 Currently, using new approaches for genetic study based on massive sequencing techniques, 90–98% of patients with aniridia can be genetically characterized.5
An update of the epidemiological and genetic aspects is presented in this paper, as well as the diagnostic algorithm to approach the molecular study of aniridia. Following the recommendations contained in this work will facilitate an adequate diagnosis for patients and their families to provide quality genetic counseling and improve the clinical management of this disease.
MethodologyDuring the preparation of this review, a search was conducted for publications on this topic indexed in Pubmed until September 30, 2020, using the following keywords: "congenital aniridia & genetics"; "PAX6 mutations", "congenital aniridia and genotype-phenotype correlation", or synonymous terms in the title, abstract, or full text fields. This search was not restricted to any temporal criteria. To determine the number of new mutations currently described in the PAX6 gene, the following databases were consulted: 1) the Human PAX6 Mutation Database, last updated on August 4, 2018 (http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6); 2) the HGMD Professional Mutation Database (version 2020.3), accessed on September 15, 2020 (https://my.qiagendigitalinsights.com/bbp/view/hgmd/pro/start.php); and 3) recent literature describing new variants not recorded in these two databases.
Epidemiological and clinical aspects of aniridiaAniridia is estimated to have an incidence of between 1:50,000–100,000 live births depending on the population studied.1 In the Spanish population, a minimum incidence of 1:135,000–238,000 has been described in the Spanish Collaborative Study of Congenital Malformations (ECEMC) using a cohort of newborns born between 1980 and 2005,6 a figure that is probably underestimated given the difficulties in diagnosing aniridia at birth. According to the latest ORPHANET report of 2020 on the prevalence of rare diseases in Europe, isolated aniridia would have an estimated incidence of 1.31/100,000.7 The incidence of WAGR syndrome is unknown5 although the risk of Wilms tumour in aniridia is estimated to be 6%.8
Although the term "aniridia" refers to the absence of iris, the iridian phenotype can vary from almost complete absence to the presence of a complete iris with minor structural alterations or pupillary abnormalities, through variable hypoplasia or iridian elongation mimicking a coloboma.5,9 Patients may also present with anterior segment anomalies throughout their lives such as: limbal dysfunction keratopathy, dry eye, Peters anomaly, juvenile glaucoma associated with alterations in the chamber angle, cataracts, ectopia lentis or lens subluxation. 90–95% of patients with aniridia exhibit variable foveal hypoplasia, and less frequently optic nerve hypoplasia or coloboma.9
Congenital aniridia may occasionally present with other systemic disorders such as reduced olfactory function, hearing loss, sleep pattern disturbances and other endocrine disorders,1 but studies with larger cohorts are needed to rule out a stochastic component thereof. Up to one third of patients may have a syndromic form.1 Patients with WAGR syndrome (MIM #194072), an acronym for Wilms’ tumour, Aniridia, Genitourinary anomalies and Developmental delay, have a 42–70% risk of developing a childhood renal tumour before the age of 10.8 Less frequent is the variant associated with obesity, called WAGRO syndrome.5 Severe iridian hypoplasia can appear in other syndromes with anterior segment dysgenesis such as Axenfeld-Rieger (MIM #180500 and MIM #601499), Peters (MIM #604229) and Gillespie (MIM #206700) syndromes, caused by genes other than PAX6.5,10 However, the presence of extraocular manifestations typical of these syndromes such as dental anomalies, cardiac anomalies, hearing loss, redundant umbilical skin or cerebellar ataxia, allows a differential diagnosis with syndromic aniridia.
Etiopathogenesis of aniridiaAniridia is mostly caused by mutations in the Paired box-6 gene (PAX6) that lead to its haploinsufficiency, i.e. to a decrease in gene dosage that does not allow the maintenance of the normal function of this transcriptional factor. PAX6 plays a fundamental role in ocular embryogenesis by regulating the spatiotemporal differentiation of the anterior and posterior segments of the eye.11 In humans, PAX6 is expressed early in the optic vesicle and later in the optic cup where it contributes to the formation of the neural and pigmentary retina.11,12 It is also expressed in ectodermal layers contributing to the differentiation of the different structures of the anterior segment, including the crystalline lens and corneal epithelium.12 In the postnatal stage, its expression is restricted to some retinal layers, lens, cornea, iris and ciliary body, where PAX6 would participate in the maintenance of these structures.12 In addition, PAX6 plays an important role in the morphogenesis and subsequent function of the central nervous system, olfactory system and pancreatic islets. Dysregulation of PAX6 expression during embryonic development, both haploinsufficiency and overexpression, leads to a wide variety of ocular anomalies in different animal models and in humans: hypoplasia of the iris, retina and optic nerve, microphthalmia, microcornea, cataracts, etc.11,12
With an extension of 22 kb, PAX6 is located in the 11p13.3 region of chromosome 11 adjacent to the WT1 gene, which is involved in the development of Wilms tumor (Figs. 1 and 2). PAX6 consists of 14 exons (11 coding) that encode a protein with two DNA-binding domains, a paired domain (PD) at the N-terminal end and a homeodomain (HD), in addition to a proline-serine-threonine (PST)-rich domain with transactivating function located at the C-TERMINAL END.11,12 In humans, 3 alternative promoters have been described that regulate gene expression in a spatiotemporal manner11,12 giving rise to several protein isoforms. The most frequent are a canonical isoform of 422 amino acids and a 5(a) isoform of 436 amino acids due to the inclusion of an alternative exon (ex5a) that interrupts the paired domain.11 The balance between both isoforms is modified throughout development, regulating the different functions that PAX6 exerts during ocular embryogenesis and postnatally.13
 The downstream regulatory region (DDR), located in the introns of the neighboring gene ELP4, is represented with the main regulatory elements that have been involved in aniridia, SIMO and E180B (in red). In addition, the breakpoints of the previously detected 11p13 deletions that have allowed us to determine the critical region for the transcriptional regulation of PAX6 are represented. Dashed blue and red lines indicate the 244 Kb region, defined by Ansari et al. and then narrowed to 18 Kb by Plaisancié et al. B) The PAX6 gene and protein are depicted. In the upper part, the exon structure is depicted with the 14 exons of the PAX6 gene, including non-coding regions (in gray) and the different coding regions, where the colors indicate the encoded protein domain, and the promoters (P0, P1 and P α). In the lower part of the image, the structure and the different functional domains of the 2 main protein isoforms, the canonical form and the isoform 5a of 422 and 436 amino acids, respectively, are represented. HD: homeodomain; PD: paired domain; PST: proline/serine/threonine-rich region.")
Representation of the PAX6 locus on chromosomal region 11p13, PAX6 gene and its regulatory elements. A) The downstream regulatory region (DDR), located in the introns of the neighboring gene ELP4, is represented with the main regulatory elements that have been involved in aniridia, SIMO and E180B (in red). In addition, the breakpoints of the previously detected 11p13 deletions that have allowed us to determine the critical region for the transcriptional regulation of PAX6 are represented. Dashed blue and red lines indicate the 244 Kb region, defined by Ansari et al. and then narrowed to 18 Kb by Plaisancié et al. B) The PAX6 gene and protein are depicted. In the upper part, the exon structure is depicted with the 14 exons of the PAX6 gene, including non-coding regions (in gray) and the different coding regions, where the colors indicate the encoded protein domain, and the promoters (P0, P1 and P α). In the lower part of the image, the structure and the different functional domains of the 2 main protein isoforms, the canonical form and the isoform 5a of 422 and 436 amino acids, respectively, are represented. HD: homeodomain; PD: paired domain; PST: proline/serine/threonine-rich region.

Schematic representation of the chromosomal regions involved in aniridia and WAGR/WAGRO syndrome at the 11p15.1-13 locus. The location of the 3 main genes involved, WT1, PAX6 and BDNF and the critical regions of the 11p deletions associated with WAGR syndrome, isolated aniridia and WAGRO syndrome are shown.
The transcriptional activation of PAX6 is highly regulated through a complex network of regulatory elements that modulate its gene expression in a tissue-specific manner.14,15 To date, about thirty of these evolutionarily conserved regulatory elements have been identified that are located in the introns of PAX6 and in intergenic regions upstream (5′ location) and downstream (3′ location) of the gene16 (Fig. 2). Most of them are located in a 3′ region located at about 150 Kb in introns 7–9 of the contiguous gene ELP4, which was identified thanks to the study of the breakpoints in patients with microdeletions in chromosome 11p13 in which the coding region of the PAX6 gene remained intact.17 The regulatory elements located in this region include the SIMO and E180 elements (Fig. 2) that regulate the expression of PAX6 during the development of different ocular structures.3,14,18 The direct involvement of SIMO in aniridia was confirmed by Bhatia et al., 2013 by identifying a point mutation in this element that affects the PAX6 binding site and, as a consequence, causes its transcriptional inhibition.18 These findings highlight the importance of regulatory regions in the development of aniridia.
Mutational spectrum of aniridiaIn approximately 70–80% of cases, classic aniridia is caused by coding mutations in PAX6 and by microdeletions of variable size in the 11p13-14 region among the remaining 20–25%.5 In its last update in 2018, the Human PAX6 Mutation Database lists 491 distinct variants identified in this gene. Since then, about 250 new mutations described in the literature have been identified,3,4,19–30 of which only 190 are collected in the HGMD database at the time of this review. Therefore, there is currently no completely updated registry of mutations that covers the entire mutational spectrum in PAX6.
Overall, 85% of the mutations described correspond to coding variants that create a premature termination codon (PTC): nonsense variants (40%), small insertions or deletions (30%) that alter the reading frame (frameshift) and splicing variants, i.e. located at the intron splicing sites and that affect the processing of messenger RNA (mRNA) (15%).16 All these variants would lead to PAX6 haploinsufficiency by the degradation of PTC-bearing mRNAs in a manner mediated by the nonsense mediated decay (NMD) system, a cellular control mechanism that eliminates aberrant mRNAs. Except for exons 13 and 14, PTC variants have been identified throughout the entire coding sequence, 40% of which are located in exons 5 and 6 that encode the PD protein domain (Fig. 3). Four recurrent mutations represent 21% of the total number of patients registered in the PAX mutational database1,16 (Fig. 3).
 are indicated; in pink, yellow and green, exons encoding the main functional domains of PAX6, paired (PD), homeodomain (HD) and proline-serine-threonine-rich domain (PST), respectively.")
Representation of the location of unique mutations in the gene and protein structure of PAX6. The count of variants described in PAX6, by exons, in the PAX6 Mutation Database, as of its last update in 2018, is depicted, as well as the most represented variants, including 4 recurrent mutations in exons 8, 9, 10 and 11. In grey, non-coding regions (5′ and 3′ UTRs) are indicated; in pink, yellow and green, exons encoding the main functional domains of PAX6, paired (PD), homeodomain (HD) and proline-serine-threonine-rich domain (PST), respectively.
The remaining 15% of the variants described do not give rise to a PTC, including non-synonymous mutations that lead to an amino acid change (missense) and stop loss variants. Most non-synonymous mutations are located in highly conserved residues of the PD domain, and the amino acid change alters the polarity and/or protein structure.16,31 Several studies focused on the in vitro study of the functional consequences of non-synonymous variants confirm that they reduce the binding to DNA and the transactivating function of PAX6 on target genes, leading to a loss of function.21,32 Others would give rise to anomalous proteins that act with a dominant negative effect interfering with the normal function of PAX6 or even gains in function due to the activation of target genes other than those regulated by the wild-type PAX6 protein.
In recent years, the involvement of variants in regulatory and non-coding regions of the PAX6 gene in aniridia has been described.3,20,28 Most are located in splicing sites of exons 2 and16,33 (Fig. 3), which are part of the non-coding region of the gene located 5′ from the translation start codon (5′UTR, untranslated region). In addition, variants have been identified in deep-intronic regions and synonymous variants in which the creation of alternative splicing is predicted by the creation of non-canonical splicing sites and/or the inclusion of new pseudoexons.3,20,33 The in vitro study of these non-coding variants by means of the "minigene" strategy, which uses artificial splicing vectors that include one or several exons of a gene, separated by intronic sequences, as well as functional splicing sites necessary for its expression, is key to confirm its pathogenicity regulated by deregulation of the normal splicing patterns of PAX6.3,28,33 In addition, gene variants have been identified in the regulatory elements that could alter the transcriptional expression of PAX6.3,18
On the other hand, up to 25% of cases of aniridia are caused by structural variants, i.e. insertions, deletions or duplications of DNA segments, as reflected in studies conducted in different cohorts.5,16,33–35 The most frequent are microdeletions of variable size in the 11p13-14 region that can affect the PAX6 gene or only its regulatory elements located at 3′, as well as a variable number of genes contiguous to PAX6.1,16,36 These contiguous gene deletions include at least WT1 in patients with WAGR syndrome and, in addition, the obesity-associated BDFN gene in WAGRO syndrome, located at a distance of about 700 Kb and 4.2 Mb from PAX6, respectively1,5,37 (Fig. 2). The inclusion in these deletions of the PRGG4 and LMO2 genes could modulate the appearance of intellectual disability or increase the risk of developing Wilms tumor.24,38 However, the participation in the phenotype of the more than 40 genes that could be included in these deletions is unknown. Less frequently, there may be other structural variants such as translocations, inversions or duplications of chromosome 11p13 involving PAX6 or its regulatory elements (Figs. 1 and 2) that have been associated with ocular and systemic abnormalities.39,40
Genotype-phenotype correlationsPAX6 variants lead to different clinical presentations depending on the nature of the mutation and its location in the gene.16 There is great inter- and intra-familial phenotypic variability in patients with PTC, so there are no clear genotype-phenotype correlations.9,16,41 However, it is generally accepted that mutations leading to gain or loss of a PTC are associated with a classic aniridia phenotype with iridian and macular hypoplasia (Table 1). A recent study in a cohort of 46 patients suggests possible correlations between the type of variant and the keratopathy associated with aniridia, with greater severity and earlier presentation in carriers of complete deletions of the gene and, on the contrary, milder and non-progressive forms associated with non-synonymous mutations.23,42 In contrast, this association was not observed in a cohort of 126 patients studied by Vasilyeva et al., although they do describe that carriers of deletions affecting the regulatory elements of PAX6 present milder phenotypes of foveal hypoplasia and keratopathy.22 Therefore, it would be necessary to study large cohorts of patients with more homogeneous clinical evaluations to confirm or rule out these observations.
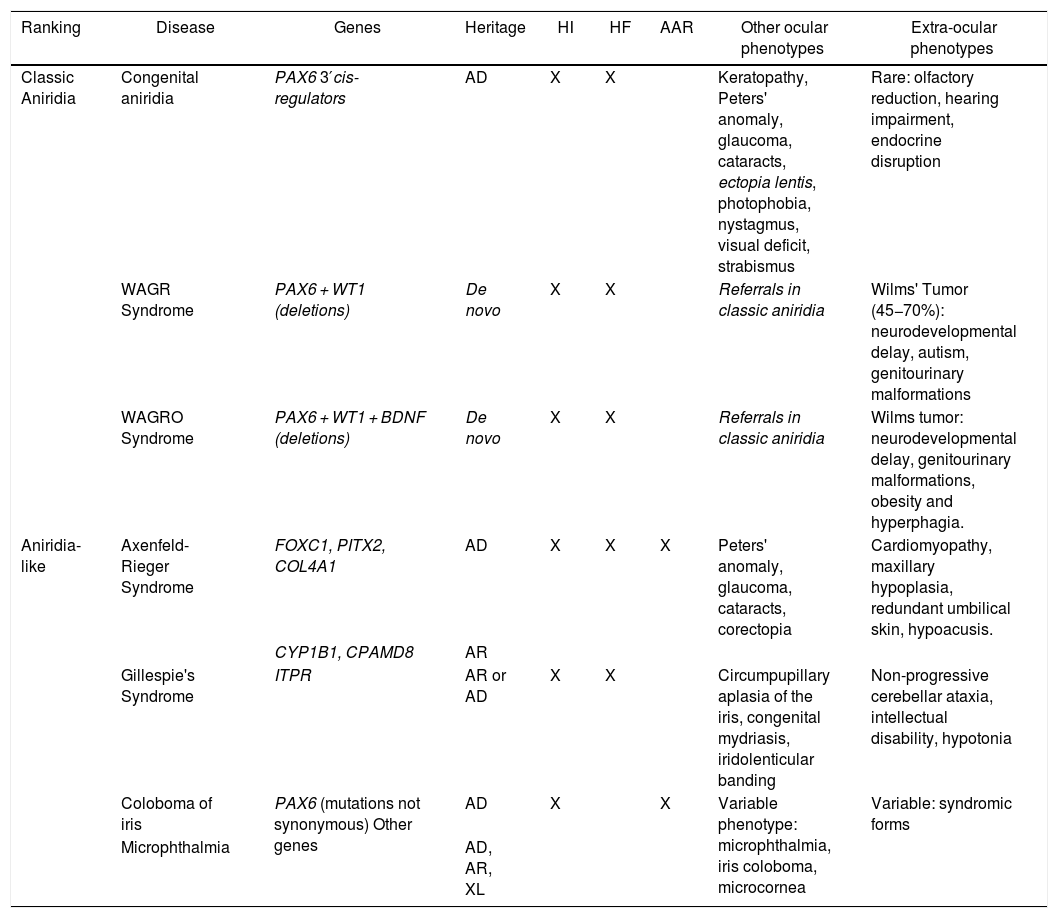
Phenotype-genotype classification of forms of aniridia and differential diagnosis with other ocular forms.
| Ranking | Disease | Genes | Heritage | HI | HF | AAR | Other ocular phenotypes | Extra-ocular phenotypes |
|---|---|---|---|---|---|---|---|---|
| Classic Aniridia | Congenital aniridia | PAX6 3′cis-regulators | AD | X | X | Keratopathy, Peters' anomaly, glaucoma, cataracts, ectopia lentis, photophobia, nystagmus, visual deficit, strabismus | Rare: olfactory reduction, hearing impairment, endocrine disruption | |
| WAGR Syndrome | PAX6 + WT1 (deletions) | De novo | X | X | Referrals in classic aniridia | Wilms' Tumor (45−70%): neurodevelopmental delay, autism, genitourinary malformations | ||
| WAGRO Syndrome | PAX6 + WT1 + BDNF (deletions) | De novo | X | X | Referrals in classic aniridia | Wilms tumor: neurodevelopmental delay, genitourinary malformations, obesity and hyperphagia. | ||
| Aniridia-like | Axenfeld-Rieger Syndrome | FOXC1, PITX2, COL4A1 | AD | X | X | X | Peters' anomaly, glaucoma, cataracts, corectopia | Cardiomyopathy, maxillary hypoplasia, redundant umbilical skin, hypoacusis. |
| CYP1B1, CPAMD8 | AR | |||||||
| Gillespie's Syndrome | ITPR | AR or AD | X | X | Circumpupillary aplasia of the iris, congenital mydriasis, iridolenticular banding | Non-progressive cerebellar ataxia, intellectual disability, hypotonia | ||
| Coloboma of iris | PAX6 (mutations not synonymous) Other genes | AD | X | X | Variable phenotype: microphthalmia, iris coloboma, microcornea | Variable: syndromic forms | ||
| Microphthalmia | AD, AR, XL |
AAR: Axelfeld-Rieger anomaly; AD: autosomal dominant; AR: autosomal recessive; FH: foveal hypoplasia; HI: iris hypoplasia; XL: X-linked.
On the other hand, it is difficult to predict the phenotype associated with non-synonymous mutations.16,41 These are identified in patients with both mild forms with partial manifestation of the phenotypic spectrum (isolated hypoplasia of the iris, fovea or optic nerve)9,27,31,43 and severe forms of classic aniridia22 (Table 1). In addition, they can be associated with isolated or syndromic phenotypes of colobomatous microphthalmia.4,21,31,43 A recent study describes that severe presentations of syndromic microphthalmia associated with PAX6 appear in carriers of nonsynonymous mutations that alter the interaction of the PD domain of PAX6 with the transcription factor SOX2, whose mutations lead to a very similar phenotype.21
Genetic diagnosis in aniridiaGenetic studies in aniridia allow us to confirm clinical suspicion, perform differential diagnosis in cases with doubtful clinical manifestations, determine the risk of developing Wilms tumour in sporadic cases of newborns or in early childhood diagnosed with aniridia, provide genetic counselling, establish the evolution and prognosis as far as possible, depending on the type of mutation, and open the possibility of performing prenatal and/or preimplantation genetic diagnosis.1,2,44 The recommendations and considerations prior to performing a genetic study in aniridia and WAGR syndrome can be found in 2 previously published guidelines for the genetic study of aniridia and WAGR syndrome.2,45 The genetic diagnosis of aniridia should include a targeted study of the PAX6 gene to screen for different types of variants depending on the inheritance and age of the patient2,44 (Fig. 4).
. The main objective is to rule out mutations and/or deletions that cause PAX6 haploinsufficiency in cases with suspected aniridia and with classic phenotype, in addition to a differential diagnosis with other genes, mainly anterior segment dysgenesis, in cases of aniridia with atypical presentations. Priority is given to the study of copy number variations in chromosome 11p in sporadic cases younger than 10 years and/or with presentation of extraocular symptoms, characteristic of WAGR syndrome (Wilms tumor, neurodevelopmental delay, genitourinary anomalies), by means of different molecular techniques, or cytogenetic techniques. Screening for PAX6 mutations will be prioritized in familial or sporadic cases older than 10 years due to its higher yield. In this algorithm we incorporate the use of NGS techniques (panels directed to PAX6 and other genes associated to ocular malformations, clinical exomes, whole exomes or genomes) to perform both analyses, always taking into account the sensitivity limitations of each technique in the evaluation of the genetic test.")
Flowchart of the genetic diagnosis of aniridia proposed for genetic diagnosis in aniridia. A diagnostic algorithm is proposed based on clinical suspicion and patient characteristics (age and family history). The main objective is to rule out mutations and/or deletions that cause PAX6 haploinsufficiency in cases with suspected aniridia and with classic phenotype, in addition to a differential diagnosis with other genes, mainly anterior segment dysgenesis, in cases of aniridia with atypical presentations. Priority is given to the study of copy number variations in chromosome 11p in sporadic cases younger than 10 years and/or with presentation of extraocular symptoms, characteristic of WAGR syndrome (Wilms tumor, neurodevelopmental delay, genitourinary anomalies), by means of different molecular techniques, or cytogenetic techniques. Screening for PAX6 mutations will be prioritized in familial or sporadic cases older than 10 years due to its higher yield. In this algorithm we incorporate the use of NGS techniques (panels directed to PAX6 and other genes associated to ocular malformations, clinical exomes, whole exomes or genomes) to perform both analyses, always taking into account the sensitivity limitations of each technique in the evaluation of the genetic test.
In cases of sporadic aniridia in children under 9 years of age with no history of ocular disease in the parents, a genetic study to rule out microdeletions in 11p13 involving the WT1 gene is a priority in order to determine the predisposition to develop Wilms tumor2,44,45 (Figs. 2 and 4). For this purpose, there are different molecular and cytogenetic approaches, with the multiplex ligation-dependent probe amplification (MLPA) technique standing out for its speed and low cost. There are commercial MLPA designs that analyze microdeletions and other structural variants in PAX6, its regulatory regions, WT1 and other genes located on 11p13-14.1,2 However, this approach does not allow to narrow down neither the size nor the exact breakpoints of the deletions. For this purpose, we recommend comparative genomic hybridization arrays (aCGH), both commercial and those specifically designed to analyze the WAGR locus2,34 which are enriched with a greater number of probes in this region, thus allowing more precise size delineation. In general, these molecular techniques can detect microdeletions in 15–25% of patients, mainly in sporadic cases.5,22,33 However, they cannot detect inversions, translocations or balanced rearrangements, so their use does not invalidate the usefulness of classical cytogenetic techniques, such as high-resolution karyotyping and FISH in patients not genetically characterised after the use of molecular techniques·
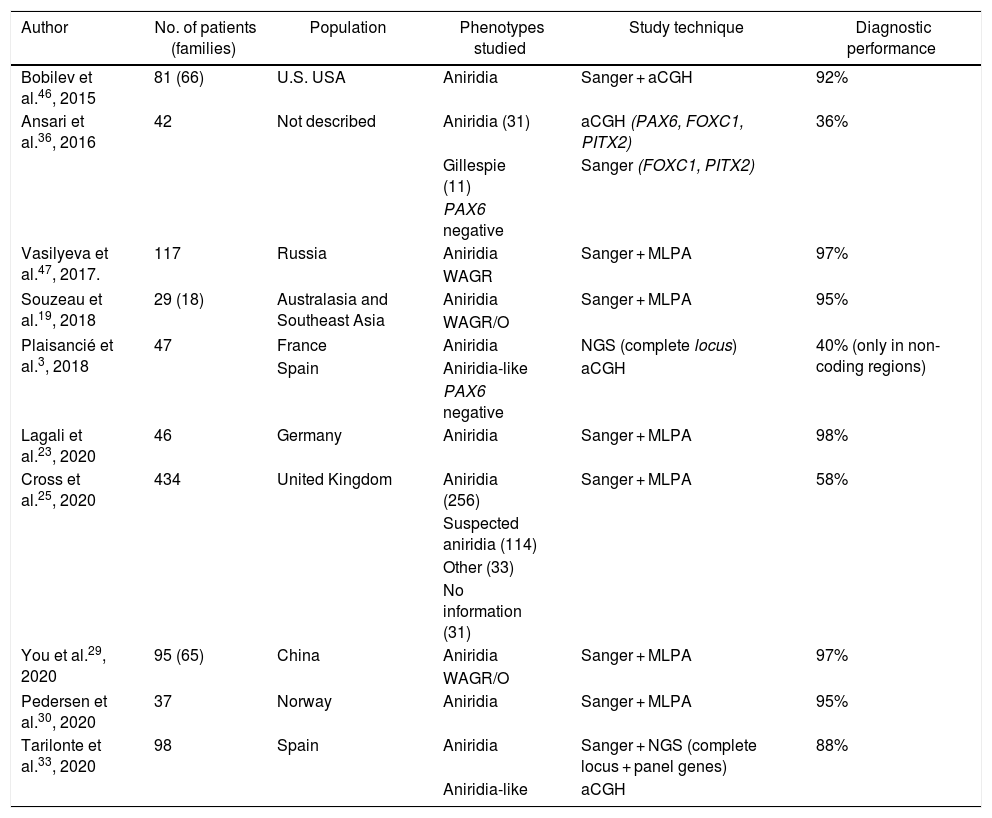
Molecular diagnosis of point variants in the PAX6 sequenceIn familial and sporadic cases not carrying microdeletions on 11p13-14, priority should be given to the study of point mutations in the coding sequence of PAX6, i.e. exons 4-142,44 (Fig. 1). The most widely used technique to date is Sanger sequencing with a detection yield between 50–90% depending on the cohort under study and the PAX6 regions analyzed (Table 2).1,22,25,30,33,44,46,47
Cohorts of patients with aniridia in different populations.
| Author | No. of patients (families) | Population | Phenotypes studied | Study technique | Diagnostic performance |
|---|---|---|---|---|---|
| Bobilev et al.46, 2015 | 81 (66) | U.S. USA | Aniridia | Sanger + aCGH | 92% |
| Ansari et al.36, 2016 | 42 | Not described | Aniridia (31) | aCGH (PAX6, FOXC1, PITX2) | 36% |
| Gillespie (11) | Sanger (FOXC1, PITX2) | ||||
| PAX6 negative | |||||
| Vasilyeva et al.47, 2017. | 117 | Russia | Aniridia | Sanger + MLPA | 97% |
| WAGR | |||||
| Souzeau et al.19, 2018 | 29 (18) | Australasia and Southeast Asia | Aniridia | Sanger + MLPA | 95% |
| WAGR/O | |||||
| Plaisancié et al.3, 2018 | 47 | France | Aniridia | NGS (complete locus) | 40% (only in non-coding regions) |
| Spain | Aniridia-like | aCGH | |||
| PAX6 negative | |||||
| Lagali et al.23, 2020 | 46 | Germany | Aniridia | Sanger + MLPA | 98% |
| Cross et al.25, 2020 | 434 | United Kingdom | Aniridia (256) | Sanger + MLPA | 58% |
| Suspected aniridia (114) | |||||
| Other (33) | |||||
| No information (31) | |||||
| You et al.29, 2020 | 95 (65) | China | Aniridia | Sanger + MLPA | 97% |
| WAGR/O | |||||
| Pedersen et al.30, 2020 | 37 | Norway | Aniridia | Sanger + MLPA | 95% |
| Tarilonte et al.33, 2020 | 98 | Spain | Aniridia | Sanger + NGS (complete locus + panel genes) | 88% |
| Aniridia-like | aCGH |
The recent implementation of Next Generation Sequencing (NGS) techniques makes it possible to analyse a large number of DNA regions in parallel and thus perform studies more cost-effectively and rapidly.5 At present, these NGS techniques represent the first approach in our laboratory for the study of aniridia and related diseases.3,4,33 The use of customised designs ("aniridia panel") allows sequencing of the entire PAX6 locus, including exons, UTR, introns and regulatory regions,3 which achieves a diagnostic yield of 85–95%,3,5,33 higher than that obtained using commercial approaches based on clinical exome sequencing. On the other hand, the decrease in the cost of NGS sequencing and the improvement in bioinformatics analysis has made it possible in some laboratories to sequence the Whole Exome Sequencing (WES)26,28 and even the entire genome Whole Genome Sequencing (WGS)48 in patients with aniridia. The latter approach covers non-coding regions and has a higher sensitivity for detecting structural variations. Depending on the design and the bioinformatics analysis performed, these NGS approaches could simultaneously detect specific and structural variants3 which will allow the study of aniridia to be addressed in the near future with a single genetic test.
Differential diagnosisDespite the congenital nature of aniridia, obtaining an early clinical diagnosis can be difficult due to the high variability in its early clinical manifestations and its high degree of overlap with other eye diseases (Table 1). In the case of infants with photophobia, nystagmus and suspected low vision in whom it is difficult to perform an ophthalmologic evaluation, genetic studies allow a differential diagnosis with other diseases with overlapping symptoms: iridian coloboma, ocular albinism, foveal hypoplasia, congenital cataracts, congenital sclerocornea and especially with anterior segment dysgenesis that can be associated with severe iridian hypoplasia5,10 (Fig. 4 and Table 1).
Some patients with mild aniridia or aniridia-like phenotype not carrying mutations in the PAX6 gene may have variants in iridodysgenesis genes associated with both dominant (FOXC1, PITX2 and COL4A1)10,36 and recessive forms (CYP1B1 and CPAMD8)10,49 (Table 1). In patients with circumpupillary aplasia of the iris, dominant and recessive mutations in the ITPR1 gene responsible for Gillespie syndrome, characterized by non-progressive cerebellar ataxia and intellectual disability, must be ruled out5 (Table 1). The use of gene panels containing these genes and others involved in overlapping diseases of ocular malformations has allowed us to identify new mutations and/or structural variants and to describe a new phenotypic association with the CRYAA cataract gene.33,49,50 Therefore, we recommend differential diagnosis using extended aniridia panels in cases with atypical phenotypes and especially in those cases without mutations in PAX633 (Fig. 4).
Inheritance and genetic counseling in aniridiaClassic aniridia is transmitted with an autosomal dominant inheritance pattern, i.e. only one allele must be altered for the ocular problem to become evident. There is usually familial segregation with several generations of affected individuals in two thirds of cases. The risk of transmission from an affected person to their offspring regardless of sex is 50% (Fig. 4).
One third of cases are sporadic due to the occurrence of a de novo variant that is usually absent in unaffected parents, but confirmation by segregation study is highly recommended. Although the risk of recurrence is very low after the birth of a sporadic case, the possibility of germline mosaicism in either parent for an apparently de novo mutation cannot be completely ruled out. Recently, we have described for the first time the presence of parental mosaicism in several families with PAX6 mutations.4,27,33 When somatic or gonosomal mosaicism is suspected in a family, quantitative analysis of the mutation using digital PCR techniques in tissues of different embryonic origin can be useful for calculating the risk of recurrence.4 On the other hand, in the case of a de novo case, a small but real risk of recurrence is established for future pregnancies so prenatal follow-up is recommended if feasible by noninvasive prenatal diagnosis (NIPD).
In the case of carriers of 11p13-14 microdeletions, these are usually associated with de novo events on the paternal chromosome,35 but it is still advisable to perform karyotyping to rule out balanced rearrangements on 11p13.
Prenatal and preimplantational diagnosisThe identification of the genetic cause in a patient with aniridia opens the possibility of performing prenatal and preimplantational studies in the family to prevent the transmission of a familial mutation or, in the case of apparently de novo mutations, of a possible germline mosaic. For this, it is necessary to carry out adequate genetic counselling where the most appropriate options are assessed according to the type of mutation, reproductive situation, age of the parents and the technical feasibility to detect the variant.2 Prenatal diagnosis is usually performed with invasive approaches based on chorionic villus biopsy or amniocentesis.44 At present it is possible to perform NIPD through the study of fetal DNA in maternal blood, indicated for de novo or paternally inherited mutations.
Future perspectivesDespite major advances in the understanding of the etiopathogenic mechanisms of aniridia, there are still many unresolved questions regarding genotype-phenotype correlations and the large phenotypic variability caused by similar mutations in the PAX6 gene. Given the complexity in the transcriptional regulation of PAX6 and its involvement in multiple spatiotemporal morphogenic networks during ocular development, the influence of regulatory elements or gene expression modulators that might contribute in part to the variable expressivity in aniridia is unknown. Somatic mosaicism could also explain some cases of intrafamilial phenotypic variability.4 As the technical sensitivity to detect low-frequency alleles improves, we trust that the study of mosaicism can be integrated into the genetic diagnosis of aniridia in the future and thus provide better genetic counseling in cases with de novo mutations. Another aspect to take into account is the characterization of cases without PAX6 mutations. The use of new WGS technologies and other approaches will allow the study of the regulatory regions of the PAX6 gene and other genes, in addition to unmasking new cases of somatic mosaics that may have gone undetected due to current limitations in sequencing techniques.
FundingThis work has been financed with funds from the Instituto de Salud Carlos III, partially financed by European Funds for Regional Development (PI17/01164, PI20/00851, PI17/1659, Miguel Servet CPII17_00006, FI18_00123, CIBERER CB06/07/0036), by the Comunidad de Madrid (RareGenomics B2017/BMD3721, EJD-2018-PRE_BMD-9453), the Federación Española de Enfermedades Raras (FEDER) and ONCE (P12-18, P20).
Conflict of interestNo conflicts of interests were declared by the authors.
The authors would like to thank the patients with aniridia for their collaboration in the research projects, as well as the health professionals, geneticists, paediatricians, ophthalmologists and laboratory technicians who have participated in the clinical and molecular diagnosis of these patients, as well as for facilitating the collection of samples and clinical data. We are grateful for the support provided by the Spanish Aniridia Association (AEA) throughout these years, as well as to the Aniridia Europe association.
Please cite this article as: Blanco-Kelly F, Tarilonte M, Villamar M, Damián A, Tamayo A, Moreno-Pelayo MA, et al. Genética y epidemiología de la aniridia congénita: actualización de buenas prácticas para el diagnóstico genético. Arch Soc Esp Oftalmol. 2021;96(S1):4–14.
