






Introduction. Hepatorenal tyrosinemia (HT1) is a treatable, inherited, metabolic disease characterized by progressive liver failure with pronounced coagulopathy. The aim of this study is to describe the clinical, biochemical, and histopathological findings in a group of Mexican HT1 patients and their outcome.
Material and methods. Medical records of HT1 patients diagnosed between 1995 and 2011 were analyzed. The diagnosis of HT1 was confirmed by detection of succinylacetone in urine or blood.
Results.Sixteen non-related HT1 cases were analyzed. Mean age at clinical onset was 9 months, and the mean age at diagnosis was 16.3 months. Main clinical findings were hepatomegaly, splenomegaly, cirrhosis, liver failure, tubulopathy, nephromegaly, Fanconi syndrome, seizures and failure to thrive. Histopathological findings were cirrhosis, fibrosis and steatosis. The HT1 group had a mortality rate of 78%. Patients who received supportive care or nutritional treatment had a 3-year survival rate of 10%. For those who underwent liver transplantation, the 6-year survival rate was 60%. In most cases pharmacological treatment with nitisinone and special dietary products were not available. The leading causes of death were fulminant liver failure, metastatic hepatocellular carcinoma, and porphyria-like neurologic crisis. Newborn screening programs in combination with the availability of orphan drugs, proper monitoring, genetic counseling, and clinical practice guidelines are needed to enable physicians to identify the disease, delay its progression, and improve patients’ quality of life.
Conclusion. The devastating natural history of HT1 is still observed in Mexican patients because they are not diagnosed and treated during the early stages of the disease.
Tyrosine (Tyr) is a non-essential aromatic amino acid derived from phenylalanine hydroxylation. Tyr metabolism takes place mainly in the cytoplasm of hepatocytes, but also in the kidney to a minor extent.1,2 The Tyr degradation pathway includes five enzymatic reactions, and metabolic disorders have been identified in four of these steps (Figure 1). The genetic tyrosinemias cause Tyr accumulation in biological fluids and tissues. Tyrosinemia type I or hepatorenal tyrosinemia (HT1 OMIM® 276700) due to deficiency of fumarylacetoacetate hydrolase (FAH EC 3.7.1.t.) is the most severe disorder described in Tyr metabolism; biochemically, patients develop hypertyrosinemia and accumulation of succinylacetone (SA) and its precursors, fumarylacetoacetate (FAA) and maleylacetoacetate (MAA). The toxicity of these compounds is thought to be responsible for the pathogenesis and its clinical signs and symptoms; some mechanisms such as increased hepatocyte apoptosis and oxidative stress might also be involved.3–5

Tyrosine metabolic pathway. 1. Phenylalanine hydroxylase. 2. Tyrosine aminotransferase. 3. 4-hydroxyphenyl-pyruvic acid dioxygenase. 4. Homogentisic acid oxidase. 5. Maleylacetoacetic acid isomerase. 6. Fumarylacetoacetic acid hydrolase. −Disorder associated with elevated Tyr in plasma. 5-ALA: 5-aminolevulinic acid. × Inhibition of heme metabolism by succinylacetone accumulation.
The FAH gene maps to human chromosome 15q25.1, where it spans 35 kb and has 14 exons, and encodes a 419 amino acid protein. To date, 71 FAH mutations have been associated with HT1.6 The incidence of HT1 is estimated at 1:100,000 live newborns worldwide; however, in Quebec, Canada, the reported frequency is as high as 1:1846 newborns.7 The incidence in Mexico is unknown.
The phenotype of HT1 is highly variable and the severity of the disease may differ depending on the age of onset of symptoms.8 Classically, the infant develops progressive liver failure within the first months of life with pronounced coagulopathy and signs of hypophosphatemic rickets secondary to renal tubulopathy. Another complication of HT1 is the porphyria-like neurologic crises caused by inhibition of heme synthesis by SA. SA is the most potent inhibitor of the enzyme 5-aminolevulinic acid dehydratase known, resulting in increased production of 5-aminolevulinic acid9 (Figure 1).The metabolic profile is indicative of hepatocellular dysfunction, showing high amino acid plasma levels, mainly Tyr, methionine (Met), and phenylalanine (Phe). There is a marked increase in alphafetoprotein (AFP), hepatic transaminases, and prolonged clotting times. Phosphaturia, glycosuria, aminoaciduria, and increased excretion of 5-aminolevulinic and phenolic acids are common.3,10
There is no other known biochemical pathway where SA is produced; therefore, it is considered as the pathognomonic marker of HT1, and SA has become the primary marker for detecting HT1 in newborn screening programs.11 The ultimate treatment for HT1 consists of a combination of pharmacological, nutritional, and surgical interventions. The orphan drug (2-[2-nitro-4-trifluoromet hybenzoyl]-1,3-cyclohexadione), known as NTBC or nitisinone, is a potent 4-hydroxyphenylpyruvate dioxygenase inhibitor. It prevents the synthesis of the toxic metabolite and effectively reduces the risk of progressive liver failure and development of hepatocellular carcinoma, which cannot be avoided only with restricted Tyr dietary intake. Nevertheless, the maximal benefit of the drug is achieved when the treatment is started in the early stages of life. Liver transplantation can prevent the hepatic and neurological complications of HT1, but its effects on renal function are unclear.12–14
In Mexico, where universal expanded newborn screening is not mandatory, diagnosis of these patients still relies heavily on clinical suspicion and subsequent investigations. The aim of this work is to describe the clinical, biochemical, and histopathological findings in a group of Mexican patients with HT1 and their outcome.
Material and MethodsThis retrospective, descriptive study was conducted at the National Institute of Pediatrics in Mexico City. The medical records of HT1 patients diagnosed at the Inborn Errors of Metabolism and Screening Laboratory between January 1995 and December 2012 were analyzed. HT1 diagnosis was determined by urinary organic acid profile, and SA detection was performed by gas chromatography/mass spectrometry. Since 2010, quantification of SA in dried blood spots by tandem mass spectrometry and plasma amino acid quantitation by high performance liquid chromatography was also performed. None of the studied patients were screened at birth for HT1.
The data collected included demographic information, gender, age at clinical symptom onset, age at diagnosis, consanguinity, number of siblings affected by HT1 or with suggestive symptoms, and clinical and biochemical data at the time of diagnosis. When available, the histopathological findings from liver biopsies were recorded and their concordance with the definitive biochemical diagnosis was analyzed. Some records were incomplete because the patients were treated at other institutions. For the mortality rate analysis, patients were grouped into two categories according to the modality of treatment received (group 1: those who underwent liver transplantation; group 2: those who did not undergo liver transplantation).
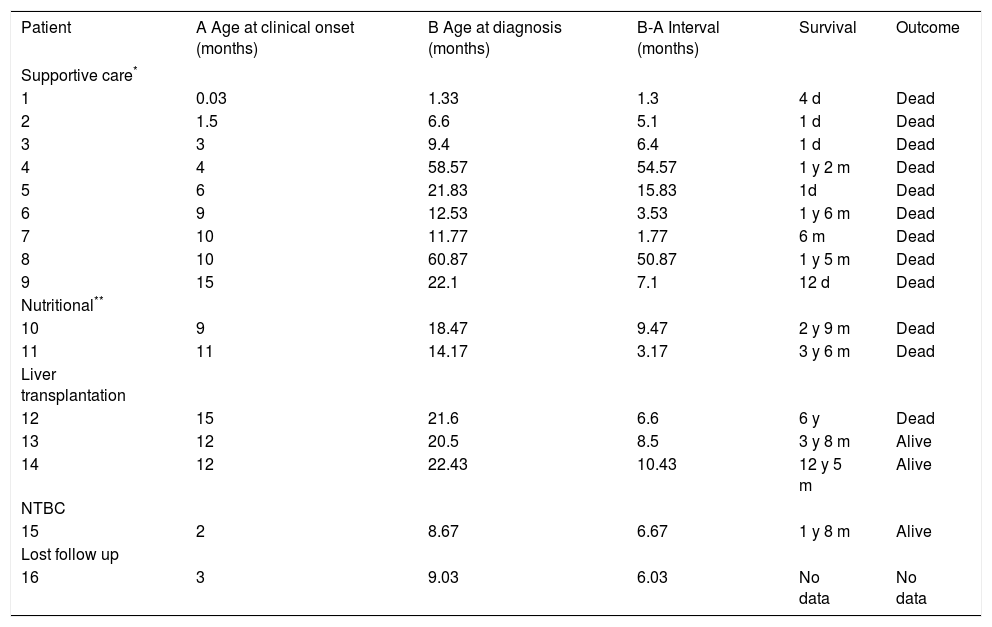
ResultsA total of 16 non-related HT1 patients from 16 families were studied, and included 7 males and 9 females. Consanguinity was documented in 3/16 families. A history of a deceased sibling with a similar clinical picture was recorded in 5 families. Clinical outcome of 15/16 patients according to type of treatment is shown in Table 1; patient 16 was lost to follow up after diagnosis. Nine patients received only supportive care (transfusions, diuretics, pain management, between others); five patients received nutritional therapy (restricted dietary Phe and Tyr) and three of these patients underwent liver transplantation. Only one patient underwent pharmacological treatment with NTBC (1 mg/kg daily) as well as Phe/Tyr dietary restriction beginning at 8 months of age. Age at clinical onset varied from 1 day in a patient who presented hypoglycemia to 15 months, with a mean of 9 months; 14/16 patients were young infants when diagnosed, and two patients were five-year-olds. The mean age at diagnosis was 16.3 months, with a minimum age of 1 month and a maximum age of 5 years. Mean time between clinical onset and definitive diagnosis was 6.6 months (Table 1).
Clinical course of HT1 Mexican patients according to their treatment.
| Patient | A Age at clinical onset (months) | B Age at diagnosis (months) | B-A Interval (months) | Survival | Outcome |
|---|---|---|---|---|---|
| Supportive care* | |||||
| 1 | 0.03 | 1.33 | 1.3 | 4 d | Dead |
| 2 | 1.5 | 6.6 | 5.1 | 1 d | Dead |
| 3 | 3 | 9.4 | 6.4 | 1 d | Dead |
| 4 | 4 | 58.57 | 54.57 | 1 y 2 m | Dead |
| 5 | 6 | 21.83 | 15.83 | 1d | Dead |
| 6 | 9 | 12.53 | 3.53 | 1 y 6 m | Dead |
| 7 | 10 | 11.77 | 1.77 | 6 m | Dead |
| 8 | 10 | 60.87 | 50.87 | 1 y 5 m | Dead |
| 9 | 15 | 22.1 | 7.1 | 12 d | Dead |
| Nutritional** | |||||
| 10 | 9 | 18.47 | 9.47 | 2 y 9 m | Dead |
| 11 | 11 | 14.17 | 3.17 | 3 y 6 m | Dead |
| Liver transplantation | |||||
| 12 | 15 | 21.6 | 6.6 | 6 y | Dead |
| 13 | 12 | 20.5 | 8.5 | 3 y 8 m | Alive |
| 14 | 12 | 22.43 | 10.43 | 12 y 5 m | Alive |
| NTBC | |||||
| 15 | 2 | 8.67 | 6.67 | 1 y 8 m | Alive |
| Lost follow up | |||||
| 16 | 3 | 9.03 | 6.03 | No data | No data |
NTBC: nitisinone. d: days. m: months. y: years.
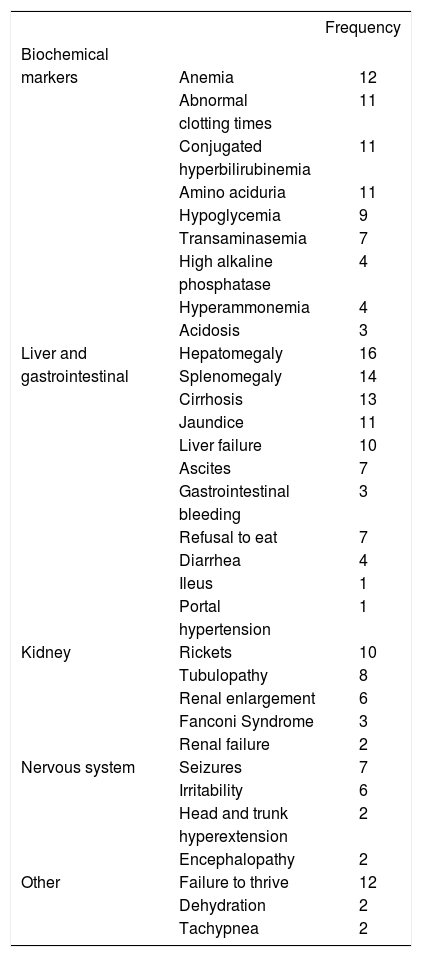
The clinical picture was characterized by progressive liver failure followed by renal and neurological abnormalities; the key biochemical markers found included anemia, abnormal clotting times, hyperbilirubinemia, aminoaciduria, and hypoglycemia (Table 2).
Biochemical markers and clinical data observed at diagnosis (n = 16).
| Frequency | ||
|---|---|---|
| Biochemical | ||
| markers | Anemia | 12 |
| Abnormal | 11 | |
| clotting times | ||
| Conjugated | 11 | |
| hyperbilirubinemia | ||
| Amino aciduria | 11 | |
| Hypoglycemia | 9 | |
| Transaminasemia | 7 | |
| High alkaline | 4 | |
| phosphatase | ||
| Hyperammonemia | 4 | |
| Acidosis | 3 | |
| Liver and | Hepatomegaly | 16 |
| gastrointestinal | Splenomegaly | 14 |
| Cirrhosis | 13 | |
| Jaundice | 11 | |
| Liver failure | 10 | |
| Ascites | 7 | |
| Gastrointestinal | 3 | |
| bleeding | ||
| Refusal to eat | 7 | |
| Diarrhea | 4 | |
| Ileus | 1 | |
| Portal | 1 | |
| hypertension | ||
| Kidney | Rickets | 10 |
| Tubulopathy | 8 | |
| Renal enlargement | 6 | |
| Fanconi Syndrome | 3 | |
| Renal failure | 2 | |
| Nervous system | Seizures | 7 |
| Irritability | 6 | |
| Head and trunk | 2 | |
| hyperextension | ||
| Encephalopathy | 2 | |
| Other | Failure to thrive | 12 |
| Dehydration | 2 | |
| Tachypnea | 2 | |
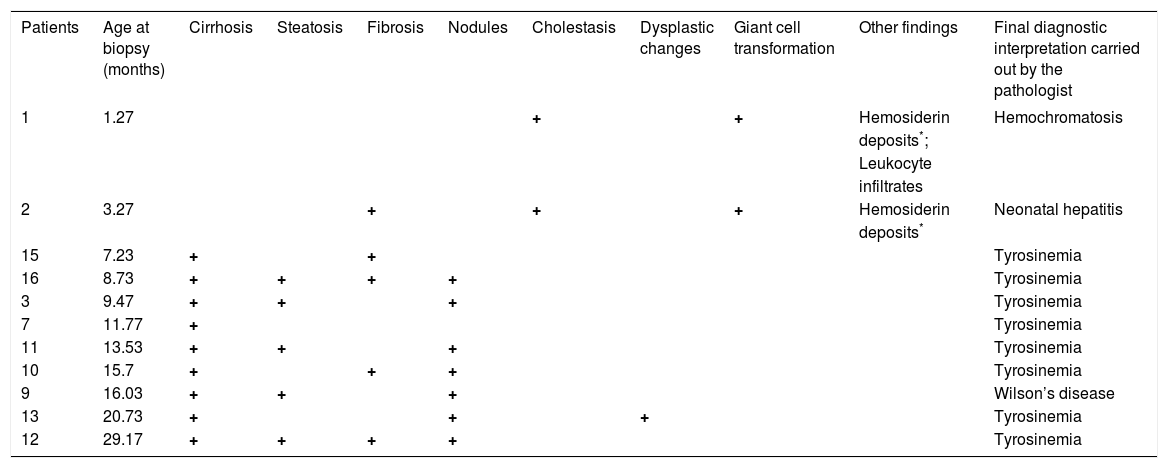
Hepatic biopsy was performed in 11/16 patients. The histopathological results were consistent with a HT1 biochemical diagnosis in 8/11 patients. The main findings were cirrhosis, fibrosis, and steatosis, and only one biopsy documented dysplastic changes. In the other 3 cases, diagnoses other than HT1 were suggested, such as neonatal hepatitis, hemochromatosis, and Wilson disease (Table 3).
Main histopathological findings of HT-1 Mexican patients.
| Patients | Age at biopsy (months) | Cirrhosis | Steatosis | Fibrosis | Nodules | Cholestasis | Dysplastic changes | Giant cell transformation | Other findings | Final diagnostic interpretation carried out by the pathologist |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.27 | + | + | Hemosiderin | Hemochromatosis | |||||
| deposits*; | ||||||||||
| Leukocyte | ||||||||||
| infiltrates | ||||||||||
| 2 | 3.27 | + | + | + | Hemosiderin | Neonatal hepatitis | ||||
| deposits* | ||||||||||
| 15 | 7.23 | + | + | Tyrosinemia | ||||||
| 16 | 8.73 | + | + | + | + | Tyrosinemia | ||||
| 3 | 9.47 | + | + | + | Tyrosinemia | |||||
| 7 | 11.77 | + | Tyrosinemia | |||||||
| 11 | 13.53 | + | + | + | Tyrosinemia | |||||
| 10 | 15.7 | + | + | + | Tyrosinemia | |||||
| 9 | 16.03 | + | + | + | Wilson’s disease | |||||
| 13 | 20.73 | + | + | + | Tyrosinemia | |||||
| 12 | 29.17 | + | + | + | + | Tyrosinemia |
The mortality rate in the HT1 group was 78%. In those patients who only received supportive treatment, the mean time of death after diagnosis was 12 days. The group of patients who received supportive or nutritional treatment had a 3-year survival rate of 10%. The 6-year survival rate of patients who underwent liver transplantation was 60% (Figure 2).

The leading causes of death were fulminant liver failure (8/11), metastatic hepatocellular carcinoma (2/11), and porphyria-like neurologic crisis (2/11).
DiscussionThe present retrospective study is the first description of a cohort of Mexican patients with HT1, documenting their clinical and biochemical findings and the causes that led to their death. Three other HT1 patients were previously reported in Mexico.15,16 Our study compiles information for 16 families, 3 with consanguinity and 5 with a precedent of a deceased sibling that can be assumed to be HT1 because of the identical clinical picture. In this cohort, hepatocellular dysfunction with coagulopathy was predominant. This hepatic syndrome was characterized by hepatomegaly, splenomegaly, cirrhosis, and liver failure accompanied by tubulopathy, nephromegaly, and Fanconi syndrome. The neurological signs presented as porphyria-like crises and were accompanied by seizures, irritability, and hyperextension of trunk and neck. Failure to thrive and rickets were also present (Table 2). The clinical picture observed in this HT1 cohort is similar to the classic description of the natural history of HT1.1,10 Contrasting with the report by Couce, et al., where symptom onset occurred between 2 and 6 months of age,17 most of the patients in our cohort reported the presenting signs after 6 months of age (9/16). However, this data does not allow us to classify the patients as having the acute or chronic form of the disease. Apparently, our cohort more resembles the chronic form of the disease (Scandinavian type); however, it is possible that the initial symptoms might have been overlooked by the parents or the family physician.
The delay in the clinical diagnosis of HT-1 patients has been recognized by other authors, despite the early onset of symptoms.17–19 In this report, we documented one case in which the interval between the onset of symptoms and diagnosis was 4.5 years (Table 1). This provides evidence that the suspicion of HT1 is difficult, even in the context of a deceased sibling. The initial clinical picture of HT1 is usually nonspecific and cannot be distinguished from other infectious or toxic hepatic syndromes. Because of its low prevalence, health personnel do not consider HT1 at first glance, leading to its being one of the last differential diagnosis options, which can cause diagnosis delay or misdiagnosis. This is one of the reasons why newborn screening is an invaluable tool for pre-symptomatic diagnosis.
HT1 is included in the newborn screening panel recommended by the American College of Medical Genetics and by the American Academy of Pediatrics, where SA is established as the primary marker of the disease.20 In contrast, the isolated use of blood Tyr has poor specificity and sensitivity because it can also be elevated in other pathologies or clinical conditions such as fasting, use of total parenteral nutrition, bleeding, transfusions, and neonatal transient hypertyrosinemia among others.1,21 In the present study, patients showed high plasma Tyr, Met, and Phe, which are secondary to the hepatic impairment.22 Tyr values were raised in all patients, and were highly variable; some patients had levels as high as 7 fold normal values, whereas others had levels that were only borderline elevated (Figure 3).

Micro- and macro-nodular cirrhosis, fibrosis, and steatosis are the main histopathological findings suggestive of HT1; however, these changes are not pathognomonic, as they are shared by a wide variety of infantile liver diseases.23–26 Especially in young children, a hepatitis-like picture appears to be nonspecific in response to several metabolic insults; therefore, HT1 diagnosis must be established based on clinical, morphological, and biochemical data. In all but 3 cases, the histopathological interpretation was consistent with the biochemical diagnosis. Of these 3, 2 showed iron storage, marked cholestasis, and giant cell transformation; however, the final diagnoses were neonatal hemochromatosis and hepatitis, respectively. Of note, biopsies were performed in the youngest patients at 1 and 3 months of age, respectively. Such findings could be present in HT1 patients, especially during earlier stages.26 The findings in the third case were reported as probable Wilson’s disease or environmental toxic cirrhosis (Table 3).
Twenty three percent of the liver biopsies were not conclusive for HT1. This confirms the robustness of the biochemical analysis, which is currently considered as the gold standard method for HT1 diagnosis.26,27 Excess SA in plasma or urine is the pathognomonic metabolite for the confirmation of HT1.1,22,28 SA can be detected in all biological fluids of untreated HT1 patients.22 The analyses must be considered part of the diagnostic approach in children with liver disease.
The mortality rate in this cohort was very high (78%) and occurred at an early age, although we observed differences in survival rates related to treatment modalities (Figure 2). Survival rates were higher in the group that underwent hepatic transplantation.
Unlike other reports where hepatocellular carcinoma is the leading cause of death,1,27 the main cause in our group was fulminant liver failure, which can be explained by the late diagnosis and the lack of specific pharmacological therapy (NTBC).
Most patients did not receive specific treatment and were only treated with supportive care. Again, we emphasize that nearly all patients were diagnosed late, when their clinical condition was so critical that some died before any therapy could be started. The 3 patients who underwent liver transplantation were treated at another Health Institute of the Mexican Ministry of Health, which has a children’s liver transplant program.29,30 The patient who received NTBC is currently aged 2 years 11 months and the pharmacological treatment resulted in correction of the coagulopathy and the anemia, and decreased SA and alpha-fetoprotein levels; however, it did not prevent the development of macronodular cirrhosis at 14 months. She is awaiting transplantation.
There is some evidence that support the benefits of NTBC administration in late diagnosed patients, but the risk of developing hepatocellular cancer remains a concern.31
Persistent urinary SA excretion following liver transplantation in HT1 patients appears to be a frequent finding and may contribute to progressive renal dysfunction in some patients.32–34
Current treatment of HT1 must be comprehensive and coupled with early diagnosis through newborn screening in order to start pharmacological treatment with NTBC as soon as possible along with nutritional therapy to control Tyr and Phe levels and, when necessary, liver transplantation.
Transplantation should be restricted to those exhibiting acute liver failure that cannot be reverted by NTBC, and in patients with suspected hepatocellular carcinoma.34 Some authors recommend that NTBC should still be administered in HT1 transplanted patients at low doses (up to 0.2 mg/kg daily) to minimize the excretion of SA in order to preserve glomerular filtration rate.32
The devastating natural history of HT1 is still observed in Mexican patients because they are not diagnosed and treated at early stages of the disease. Our results are consistent with previous reports; all patients had progressive liver failure associated with high mortality. Clearly, newborn screening programs combined with the availability of orphan drugs (NTBC), proper monitoring, genetic counseling, and clinical practice guidelines are much needed to allow physicians to suspect the disease, delay its progression, and improve the quality of life of patients. If nitisinone and Tyr/Phe restricted special formula become available for Mexican patients, especially if tyrosinemia is added to the neonatal screening panel, the outcome is expected to improve rapidly.
Abbreviations- •
HT1: hepatorenal tyrosinemia.
- •
5-ALA: 5-aminolevulinic acid.
- •
AFP: alpha-fetoprotein
- •
EC: enzyme code number
- •
FAA: fumarylacetoacetate
- •
FAH: fumarylacetoacetate gene
- •
FAH: fumarylacetoacetate hydrolase
- •
MAA: maleylacetoacetate
- •
Met: methionine
- •
NTBC 2: -[2-nitro-4-trifluoromethybenzoyl]-1,3-ciclohexadione; nitisinone.
- •
OMIM®: Online Mendelian Inheritance in Man.
- •
Phe: phenylalanine.
- •
SA: succinylacetone.
- •
Tyr: tyrosine.



