





Hepatitis A virus (HAV) infection resolves in most patients uneventfully within weeks from the onset of the disease. In rare cases, however, it may relapse or cause prolonged cholestasis. Here we present a case of a 36-year-old female patient who developed severe pruritus and jaundice three weeks after initially uncomplicated hepatitis A. A relapse of the infection was excluded. Since therapy with colestyramin, antihistaminics, naloxon and ursodeoxycholic acid (UDCA) did not improve symptoms, we decided to perform plasma absorption and to start rifampicin therapy. Under these measures, pruritus and jaundice, as well as serum bilirubin levels improved gradually and after four plasmapheresis sessions we were able to discharge the patient. Genetic testing showed the presence of two procholestatic polymorphisms, the c.3084 [GG] variant within the gene encoding the hepatocanalicular bile salt transporter ABCB11 and the c.711 [AT] variant of the phosphatidylcholine floppase ABCB4. We speculate that this compound ABCB4-ABCB11 genotype led to a severe intrahepatic cholestasis in the setting of HAV infection. In conclusion, our case suggests that polymorphisms within the hepatocanalicular transporters may contribute to a more pronounced course of HAV infection. Although dedicated studies in large cohorts of patients are needed to confirm this observation, we speculate that patients carrying procholestatic hepatobiliary transporter variants may benefit from vaccination against hepatitis A.
Hepatitis A is the most common acute viral hepatitis worldwide.1 The disease is in general self-limiting2 and in adults typically manifests with initial flu-like and gastrointestinal symptoms followed by jaundice.3 Of note, two rare variants of the hepatitis A infection, characterised by a cholestatic4 and/or biphasic course1 have been reported. The cholestatic variant is characterized by pruritus and jaundice, as well as elevation of serum bilirubin and alkaline phosphatase (AP), whereas aminotransferase activities remain normal.1 Conversely, in the biphasic course, the infection relapses a few weeks after the initial phase.5 The latter stage of the disease is characterised by increase of aminotransferase levels and presence of hepatitis A virus in patients stool.
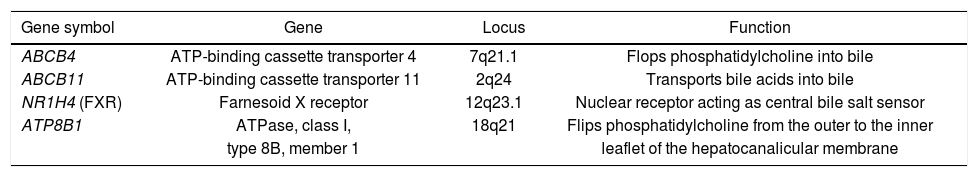
Physiological bile secretion, necessary for bile flow through the biliary tract, is maintained by a set of hepatocanalicular proteins transporting major biliary lipids from hepatocytes into the bile canaliculi.6 In short, these ATP-binding-cassette (ABC) proteins, namely ABCB4, ABCB11 and ABCG5/8, transport phosphatidylcholine, bile salts and sterols, respectively (Table 1). The composition of phospholipids within the hepatocanalicular membrane is maintained by ATP8B1, whereas the nuclear receptor FXR regulates the expression of enzymes and transporters involved in bile salt metabolism. Previous genetic studies demonstrated that dysfunction of these transport proteins results in cholestatic disorders.7,8 Indeed, several common and rare variants within the transports have been identified as genetic risk factors for cholestasis.9,10 Of note, in many cases the cholestatic phenotype is driven by an interaction between genetic predisposition and environmental modulators.11 In other words, carriers of the risk variants develop cholestasis in the presence of additional triggers affecting the expression or function of the transporters destabilizing the liver function (e.g. drugs, hormones,12 or infections13,14); otherwise no specific liver phenotype is observed.
List of procholestatic candidate genes.
| Gene symbol | Gene | Locus | Function |
|---|---|---|---|
| ABCB4 | ATP-binding cassette transporter 4 | 7q21.1 | Flops phosphatidylcholine into bile |
| ABCB11 | ATP-binding cassette transporter 11 | 2q24 | Transports bile acids into bile |
| NR1H4 (FXR) | Farnesoid X receptor | 12q23.1 | Nuclear receptor acting as central bile salt sensor |
| ATP8B1 | ATPase, class I, | 18q21 | Flips phosphatidylcholine from the outer to the inner |
| type 8B, member 1 | leaflet of the hepatocanalicular membrane |
Here we present a case of a young female patient with acute hepatitis A infection who developed severe cholestasis with intractable pruritus three weeks after the initially uneventful infection. These symptoms could be alleviated only with rifampicin and plasmapheresis. Genotyping of procholestatic transpoter variants revealed that the patient carried a combined ABCB4-ABCB11 risk genotype, which contributed to severe and prolonged cholestasis in the setting of acute hepatitis A.
Case ReportA 36-year-old Caucasian female, with no significant past medical history, was referred to our unit with acute HAV infection. The patient was a mother of two children and at the time of admission she was under a combined oral contraceptive therapy (dienogest 2 mg/day plus ethinylestradiol 30 micrograms/ day). Alcohol abuse was denied, and none of the pregnancies was complicated by cholestasis. The laboratory tests revealed that the patient was positive for IgM anti-HAV antibodies; other liver diseases were excluded, in particular HBV, HCV, CMV and EBV infections, AIH, PBC, Wilson disease and hemochromatosis. Our patient carried no protective antibodies against HBV (HBsAg negative, anti-HBs/ anti-HBc negative). After an initially uncomplicated course (maximal ALT = 1339 U/I [Figure 1, 1st. phase]) the patient was discharged. However, three weeks afterwards she was readmitted with severe pruritus, progressive jaundice, and nausea. The pruritus was so severe that it led to sleep deprivation and excoriations due to aggressive scratching. As shown in Figure 1 (2nd. phase), laboratory tests revealed increased serum bilirubin levels (26.2 mg/dL), but normal gamma-GT, aminotransferases and AP activities. Anti-HAV-IgG antibodies were detected but hepatitis A virus in stool was absent. Abdominal ultrasound performed after an overnight fast demonstrated a contracted gallbladder consistent with decreased bile secretion; biliary obstruction was excluded. Liver histology (Figure 2) showed parenchymal inflammation and cholestasis but no liver fibrosis. To alleviate the severe and progressive pruritus we initiated a sequential therapy with ursodeoxycholic acid (UDCA), colestyramin, antihistaminic drugs (i.e. dimetidene) and naloxon without any effect on itching. Hence we initiated the plasma absorption (PAP) with Adsorber Plasorba BR-350 and oral therapy with rifampicin (600 mg once daily). Laboratory tests and pruritus gradually improved under these measures. In total, four consecutive PAP sessions of plasma absorption were performed in 14 days. No adverse effects occurred. As shown in Figure 1, liver tests almost normalised, and the patient could be discharged. During the subsequent 2 years follow-up the patient remained healthy.
 improved dramatically once plasma absorption was initiated. Aminotransferase activities (continued line) remained normal throughout the latter phase of the disease.")
 from the biopsy performed during the 2nd. cholestatic phase, phase of the disease. The hepatic tissue was characterised by parenchymal inflammation and intrahepatic cholestasis, however no signs of fibrosis were detected.")
To detect genetic factors contributing to severe phenotype, we genotyped procholestatic mutations and polymorphisms in the ABCB4 (p.R590Q, c.711A>T), ABCB11 (p.E297G, p.A444V, p.D482G, c.3084A>G), ATP8B1 (p.N45T, p.E429A, p.I661T) and FXR (c.-1 G>T) genes. The genotyping was performed using PCR-based assays with 5'-nuclease and fluorescence detection (TaqMan). The variants ABCB11 p.E297G and APT8B1 p.N45T were sequenced. We detected two procholestatic variants within the ABCB4 and ABCB11 genes. The patient carried the common exonic ABCB11 (GG) variant c.3084A>G (rs497692)15-19 which leads to skipping of exon 24,20 and was heterozygous for the synonymous ABCB4 SNP c.711A>T (rs2109505), previously associated with an increased risk of intrahepatic cholestasis of pregnancy.9,12
DiscussionAn estimated 1.5 million of clinically apparent HAV infections occur yearly worldwide,21 and most of these patients recover uneventfully.22 Here we present a course of HAV infection which triggered intractable cholestasis in a patient carrying a combined procholestatic risk genotype. Indeed, previous reports13,14 have demonstrated that infections may trigger cholestatic disorders in genetically predisposed individuals. Since we excluded other hepatic disorders, hepatitis A seems to be the major trigger of cholestasis, however the pronounced symptoms may be, at least in part, explained by the presence of two cholestatic ABCB4 and ABCB11 transporter variants. The transcription and splicing analyses by Byrne, et al.20 showed that the ABCB11 c.3084A>G polymorphism, previously associated with primary biliary cirrhosis,18 intrahepatic cholestasis of pregnancy,15 and benign recurrent intrahepatic cholestasis,16 promotes skipping of exon 24, eventually leading to dysfunction of the hepatobiliary bile salt export pump.20 Moreover, the presence of heterozygosity for the ABCB4 SNP further increases the risk of cholestasis.9,12 We speculate that in the setting of HAV infection this specific ABCB4-ABCB11 genotype was the driving force for severe cholestasis. Of note, we excluded the so-called biphasic course of hepatitis A infection as HAV was not detectable at the time of the second admission. For the same reason the patient did not receive steroids, which have been postulated5,23 as therapeutic option in patients with recurrent hepatitis A infection as well as with the cholestatic course or secondary autoimmune hepatitis.24
According to the latest recommendations for treating cholestatic pruritus,25 we introduced several drugs sequentially, which however did not affect the itching. Hence, we decided to perform plasma absorption and to initiate a therapy with rifampicin, which eventually lowered the bilirubin levels and alleviated the symptoms. Rifampicin has for long been used in the treatment of cholestatic pruritus,26 but only latest studies27 have identified the mechanism of its function. In short, rifampicin exerts it effects by inducing hepatic expression of the bilirubin conjugating enzyme UGT1A1 (UDP glucuronosyltransferase 1 family, polypeptide A1) and the multispecific organic anion transporter ABCC2.27 Moreover rifampicin increases the expression of CYP3A4, which induces the detoxification of bile acids.27 On the other hand, extracorporal liver support systems can be used in patients with pruritus and increased serum bilirubin levels. We chose a plasma absorption column, which allows absorption of bilirubin and other potentially toxic substances, namely bile acids and aromatic amino acids.28 Although others have reported hemodynamic instability, major bleedings or clotting as serious but rare complications of the treatment,29 none of these in our patient.
To our knowledge this is the first report showing a possible association between procholestatic ABCB4 and ABCB11 variants and severe cholestasis following hepatitis A. Of note, the prevalence of the ABCB11 [GG] risk variant reaches 20-50% in the European populations.30 Moreover, the ABCB4 c.711 risk variant is caused by the major [A] allele. Hence this genotype, composed of two common alleles in different loci, is present in a relatively large number of individuals, who once infected with HAV may be at a risk of severe course of the disease. Although genotyping of further patients is necessary to confirm the association between the hepatocanalicular transporter variants and severe cholestasis in the setting of hepatitis A, it can be speculated that carriers of the at-risk genotypes would benefit from HAV vaccination.
Abbreviations- •
HAV: hepatitis A virus.
- •
AP: alkaline phosphatase.
- •
ABCB4: ATP-binding cassette transporter 4.
- •
ABCB11: ATP-binding cassette transporter 11.
- •
ABCG5/8: ATP-binding cassette transporter 5/8.
- •
ATP8B1: ATPase, class I, type 8B, member 1.
- •
FXR: farnesoid X receptor.
- •
HBV: hepatitis B virus.
- •
HCV: hepatitis C virus.
- •
CMV: cytomegalovirus.
- •
EBV: Epstein-Barr virus.
- •
ALT: alanine aminotransferase.
- •
AIH: autoimmune hepatitis.
- •
PBC: primary biliary cirrhosis.
- •
UDCA: ursodeoxycholic acid.
- •
PAP: plasma absorption.
- •
ABCC2: multispecific organic anion transporter.
We declare that we have no conflict of interest.
Financial DisclosureWe declare that we have nothing to disclose.



