






Background and rationale. The REPLACE study (NCT01571583) investigated telaprevir-based triple therapy in patients who have recurrent genotype 1 hepatitis C virus (HCV) infection following liver transplantation and are on a stable immunosuppressant regimen of tacrolimus or cyclosporin A. Patients received telaprevir 750 mg 8-hourly with pegylated interferon 180 μg weekly and ribavirin 600 mg daily, followed by a further 36 weeks of pegylated interferon and ribavirin alone and 24 weeks of follow-up. Efficacy (sustained virological response [SVR] 12 weeks after last planned study dose), safety and tolerability of telaprevir throughout the study were assessed. Pharmacokinetics of telaprevir, tacrolimus and cyclosporin A were also examined.
Results. In total, 74 patients were recruited. Overall, 72% (53/74; 95% CI: 59.9 to 81.5) of patients achieved SVR at 12 weeks following completion of treatment. Anticipated increases in plasma concentrations of tacrolimus and cyclosporin A occurred during telaprevir treatment and were successfully managed through immunosuppressant dose reduction and, for tacrolimus, reduced dosing frequency. Safety and tolerability of telaprevir-based triple therapy were generally comparable with previous data in non-transplant patients, although rates of reported anemia (55% [41/74]) were higher. Elevated plasma creatinine (46% [34/74]) was observed during REPLACE - consistent with the post-liver transplant population and the co-administered immunosuppressants.
Conclusion. Telaprevir-based triple therapy in patients with recurrent genotype 1 HCV infection following liver transplantation produced high rates of SVR. Therapeutic concentrations of immunosuppressants were maintained successfully through dose modification during telaprevir treatment.
Hepatitis C virus (HCV) remains the primary indication for liver transplantation in Europe.1 HCV infection almost invariably recurs in patients who have detectable levels of HCV RNA prior to transplant2,3 and sets the stage for accelerated fibrosis and dysfunction of the graft'; cirrhosis related to HCV occurs in 20-30% of graft recipients within 5 years of the transplant.4–6 Treatment of recurrent HCV in liver transplant patients aims to prevent complications that can lead to graft loss.7,8
After the discovery of the hepatitis C virus in 1989,9 development of new therapies was slow with only small increases in SVR rates for over 20 years.10 Commonly used dual therapy of pegylated interferon (Peg-IFN, P) in combination with ribavirin (RBV, R) achieves sustained virological response (SVR) rates as low as 40-50% in HCV genotype 1 patients.10 In patients who have undergone liver transplantation, SVR rates are even lower with rates of 23-31%.11 In 2011, adding the first-wave direct-acting antivirals (DAAs) telaprevir (TVR) or boceprevir (BOC) to PR resulted in marked improvements in SVR rates compared with dual therapy.12–17 HCV drug treatment rapidly evolved, and it was only three years later that the secondwave of drugs began to be approved for the treatment of HCV. These include triple therapy of simeprevir (SMV) or sofosbuvir (SOF) with PR, or interferon-free therapy of sofosbuvir in combination with simeprevir, ledipasvir (LDV) or daclatasvir (DCV).18–21 More recently, the combination of ritonavir-boosted paritaprevir (PTV/r), ombitasvir (OBV) and dasabuvir (DSV) has become available.22,23
Although these interferon-free second-wave therapies are now becoming the standard of care for treating HCV, the close proximity of approvals of first-wave DAAs to second-wave DAAs means their clinical development programs have overlapped.
Since cyclosporin A (CsA) and tacrolimus (TAC) are both substrates for both cytochrome P450 3A4 and P-glycoprotein, treating post-transplant patients with DAAs may be challenging.24–26 TVR and BOC are both substrates and inhibitors of cytochrome P450 3A4 (TVR is also a substrate and inhibitor of P-glycoprotein), so there is a potential for drug interactions upon co-administration with these immunosuppressants.16,17 Indeed, drug-drug interaction studies with TVR and BOC have shown that these DAAs can increase plasma concentrations of the immunosuppressants CsA and TAC.27–30 Drug-drug interactions with CsA and TAC and the second-wave DAAs may also need to be managed as SMV, DCV and ritonavir are also inhibitors of cytochrome P450 3A4, and SMV, SOF, DCV, LDV, PTV/r, DSV are inhibitors of P-glycoprotein.18–23 It is therefore vital that the clinical benefits of the DAAs are examined in the liver transplantation population. By managing the drug-drug interactions between TVR, TAC and CsA through immunosuppressant dose modification, a proof-of-concept study indicated that TVR-based triple therapy was viable to use in post-liver transplant patients who have experienced HCV recurrence.27,28 Following on from this, the REPLACE study described here aimed to determine the efficacy, safety, and tolerability of TVR in combination with PR in a large population of patients with chronic, genotype 1 HCV infection, who had undergone liver transplantation and were receiving TAC or CsA.
As well as providing information on TVR in transplant patients, given the lack of data in this population, the results of our study can also be used to inform the examination of future DAAs in this patient population.
ObjectivesThe main study objective was to determine the efficacy of TVR 750 mg every 8 hours (q8h) in combination with PR in patients with chronic, genotype 1 HCV infection who had undergone liver transplantation and were receiving TAC or CsA. Efficacy was assessed as the number of patients achieving SVR12, defined as having undetectable HCV RNA 12 weeks following the last planned dose of study medication.
Secondary objectives included the comparison of SVR rates in the REPLACE study with historical control rates of SVR with dual therapy, the evaluation of safety and tolerability of TVR in combination with PR and TAC or CsA, the pharmacokinetics of TVR and the immunosuppressant drugs TAC and CsA when co-administered, the comparison of pre- and post-treatment liver graft histology, and the dose requirements for TAC and CsA.
Material and MethodsStudy designIn this open-label, single-arm, multicenter, Phase IIIB study, patients received TVR 750 mg every 8 h (q8h) in combination with Peg-IFN 180 µg/week, and an initial dose of RBV 600 mg daily for 12 weeks. Doses of RBV were subsequently titrated by the investigator up to 1,0001,200 mg daily, depending on patients’ weight, tolerability, and local practice. This was followed by 36 weeks of PR treatment alone. The study comprised a 4-week screening period, 48 weeks of HCV treatment and a 24-week follow-up phase, with SVR assessed at follow-up week 12 (Supplementary Figure 1). Treatment was permanently discontinued for viral breakthrough at week 4 or 8 in patients who experienced a > 1 log increase in HCV RNA from the lowest level reached or if patients had a value of HCV RNA > 100 IU/mL when their HCV RNA levels had previously become < 25 IU/mL during treatment. At week 12, treatment was permanently discontinued if HCV RNA was > 1,000 IU/mL and at weeks 24 or 36 it was discontinued if HCV RNA was detectable. Changes in liver graft histology were evaluated through comparison of pre- and post-treatment liver graft biopsies. All patients provided written, informed consent prior to the start of the study. This study was reviewed and approved by independent ethics committees and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The REPLACE study was registered with ClinicalTrials.gov (NCT01571583).
PatientsPatients had received a first-time liver transplant > 6 months to 10 years prior to enrolling in the study, with a primary pre-transplant diagnosis of chronic, genotype 1 HCV infection that had recurred following transplantation. HCV treatment-naïve patients and patients who had been treated with PR prior to transplant were included. Only patients with METAVIR F0-3 fibrosis were enrolled, and patients with F0 fibrosis were only included if biopsy showed necroinflammation grade >2, or if alanine aminotransferase levels were > 2 times the upper limit of normal.
Exclusion criteria for this study included current infection or co-infection with non-genotype 1 HCV and having received previous treatment with a DAA for HCV. Patients who had received treatment for HCV with approved or investigational drugs following liver transplantation were not enrolled. Any patients with histological evidence of graft rejection on the most recent liver biopsy were excluded, as were patients with contraindications to treatment with PR.
Immunosuppressant treatment regimenAll patients were on a stable immunosuppressant regimen of either TAC or CsA for at least 1 month prior to the screening visit. Combination treatment of TAC or CsA with mycophenolate mofetil, and the addition of low-dose prednisone (defined as a daily dose of ≤ 5 mg) were permitted as part of the immunosuppressant regimen.
In order to minimize the risk of TAC or CsA levels becoming supra/subtherapeutic during co-administration with TVR, immunosuppressant dosing, trough concentrations, and safety parameters were assessed for the first 16 patients entering the study during the initial 4 weeks of treatment (10 receiving TAC and six receiving CsA). Results were evaluated by a Safety Monitoring Committee and used to formulate dose adjustment guidance for the participating investigators to use in the remaining patients.
The guidance based on this review was that in patients with a pre-TVR TAC dose of ≤ 5 mg daily, the suggested TAC dose was 0.2-0.5 mg with subsequent dosing every 3 to 5 days. Data on patients receiving > 5 mg daily TAC dose prior to treatment with TVR were limited.
For patients receiving 100–200 mg CsA daily prior to initiating treatment with TVR, a lower starting dose of 2550 mg CsA daily was suggested.
Following the end of TVR treatment, it was suggested that patients start a dose of TAC or CsA at least as high as their original dose prior to receiving TVR.
Efficacy of TVR-based triple therapyPlasma HCV RNA was quantified using the Roche High Pure COBAS® Taqman® HCV test 2.0, which has a lower limit of quantification of 25 IU/mL and a limit of detection of 15 IU/mL. Samples of plasma for RNA quantification were collected during screening and on day 1 (baseline), day 7, weeks 2, 3, 4, 8, 12, 16, 24, 36, and 48 during treatment and 4, 12, and 24 weeks after completion of treatment.
Viral sequencingPopulation sequencing analyses of the HCV NS3-4A protease were performed on all baseline samples and in the case of on-treatment virologic failure or relapse. HCV RNA was isolated from the plasma and the NS3-4A protease was amplified by real-time polymerase chain reaction. HCV RNA was sequenced if the values were above the limit of detection of the sequencing assay (~1,000 IU/mL). Virology analyses were performed using the list of TVR-resistant variants identified in patients enrolled in Phase II or III studies with TVR and who did not achieve a SVR.31
Pharmacokinetic analysesBlood samples for TVR pharmacokinetic analysis were taken at weeks 2, 4, 6, and 8. Samples were analysed by liquid chromatography-tandem mass spectroscopy. The analysis of TVR plasma concentration was performed centrally. The pharmacokinetic parameters of average steady-state plasma concentration (Css,avg), predose plasma concentration (Ctrough) and the area under the curve over the dosing interval (AUCt) were calculated.
Measurement of TAC or CsA plasma concentrations were performed locally so that, in combination with safety assessments, investigators could titrate the doses of TAC or CsA according to standard treatment practices. These analyses were carried out at prespecified time points as well as at the discretion of the investigator. Descriptive statistics and graphical analyses were performed to determine the effect of TVR on the plasma concentrations of TAC and CsA.
Adverse events and safetyThe safety and tolerability of TVR were monitored throughout the study. Samples for serum chemistry, hematology, coagulation, and urinalysis were collected, and local electrocardiograms were recorded at specified time points throughout the study period. Additional analyses were performed at the discretion of the investigators.
Adverse events (AEs) were reported by patients and monitored by investigators throughout the study period. The severity of AEs and their relationship to TVR were recorded.
Statistical analysesA sample size of 72 patients was considered adequate to provide 90% power to establish the superiority of the primary endpoint (SVR12) over the historical control rate of 31% when an observed SVR rate of 50% is assumed. SVR was estimated with a 95% confidence interval (CI). The historical SVR rate used in the study protocol, 27% with a 95% CI of 23 to 31, was estimated through a meta-analysis of 16 previously published studies in this post-transplant population.11 If the lower bound of the 95% CI excluded the prespecified historical control of 31%, the null hypothesis was rejected.
For continuous parameters, summary descriptive statistics included a number of observations, mean, 95% CI, standard deviation (SD), standard error (SE), minimum, median, Q1, Q3, and maximum. For categorical parameters, summary statistics included frequency and percentages.
For virological response, a non-completer was regarded as a virological failure (NC = F virological failure imputation); intermittent missing values were imputed with 1 (i.e., response) if the immediate preceding and following visits demonstrated response and with 0 (i.e., no response) otherwise.
Statistical analyses were carried out using SAS software v9.2, with the exception of the pharmacokinetic analysis, which was conducted via non-linear mixed-effects modelling with NONMEM software version VII level 2.0.
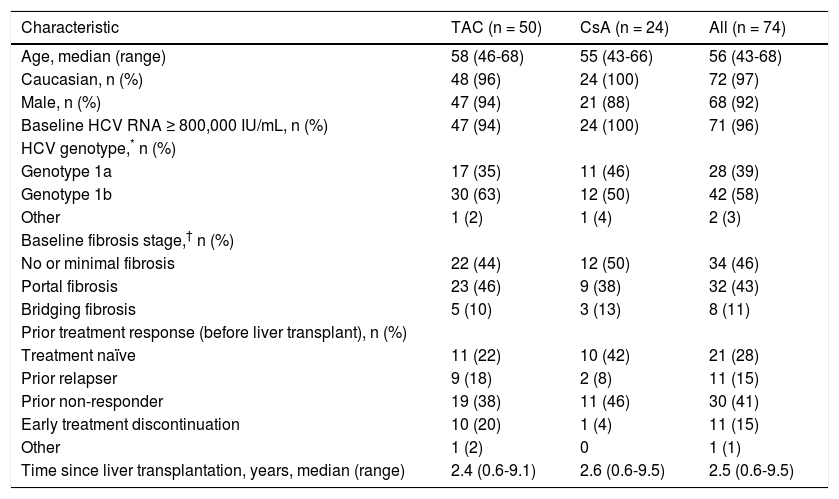
ResultsBaseline patient characteristicsIn total, 74 patients were enrolled in the study and the majority of patients were male (92%) and Caucasian (97%), with a median age of 56 years (Table 1). The majority of patients (58%) had HCV genotype 1b, and 96% of patients had HCV RNA levels ≥ 800,000 IU/mL at baseline (Table 1). Overall, 28% (21/74) of patients were HCV treatment naïve; of the remaining 53 patients who had received HCV treatment prior to transplantation, 57% (30/53) were prior relapsers, and 21% (11/53) were prior non-responders (Table 1). Patients with no or minimal fibrosis made up 46% of the patients enrolled in the study, 43% of patients had portal fibrosis, and the remaining 11% had bridging fibrosis (Table 1). Of the 74 patients enrolled in the study, 88% completed the TVR treatment phase.
Baseline patient characteristics for all patients, by immunosuppressant regimen and overall.
| Characteristic | TAC (n = 50) | CsA (n = 24) | All (n = 74) |
|---|---|---|---|
| Age, median (range) | 58 (46-68) | 55 (43-66) | 56 (43-68) |
| Caucasian, n (%) | 48 (96) | 24 (100) | 72 (97) |
| Male, n (%) | 47 (94) | 21 (88) | 68 (92) |
| Baseline HCV RNA ≥ 800,000 IU/mL, n (%) | 47 (94) | 24 (100) | 71 (96) |
| HCV genotype,* n (%) | |||
| Genotype 1a | 17 (35) | 11 (46) | 28 (39) |
| Genotype 1b | 30 (63) | 12 (50) | 42 (58) |
| Other | 1 (2) | 1 (4) | 2 (3) |
| Baseline fibrosis stage,† n (%) | |||
| No or minimal fibrosis | 22 (44) | 12 (50) | 34 (46) |
| Portal fibrosis | 23 (46) | 9 (38) | 32 (43) |
| Bridging fibrosis | 5 (10) | 3 (13) | 8 (11) |
| Prior treatment response (before liver transplant), n (%) | |||
| Treatment naïve | 11 (22) | 10 (42) | 21 (28) |
| Prior relapser | 9 (18) | 2 (8) | 11 (15) |
| Prior non-responder | 19 (38) | 11 (46) | 30 (41) |
| Early treatment discontinuation | 10 (20) | 1 (4) | 11 (15) |
| Other | 1 (2) | 0 | 1 (1) |
| Time since liver transplantation, years, median (range) | 2.4 (0.6-9.1) | 2.6 (0.6-9.5) | 2.5 (0.6-9.5) |
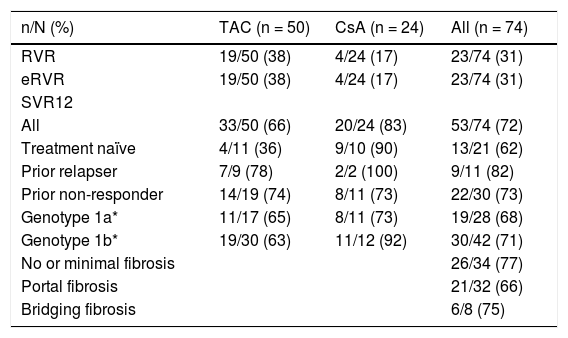
The primary endpoint of SVR12 was achieved by 72% (53/74, 95% CI: 59.9 to 81.5) of all patients enrolled in the study, 66% (33/50, 95% CI: 51.2 to 78.8) of patients receiving TAC, and 83% (20/24, 95% CI: 62.6 to 95.3) of patients receiving CsA achieved SVR12 (Table 2). The difference in SVR rates between TAC and CsA is not significant given the small sample size and lack of randomization to immunosuppressant. Rates of efficacy by prior response to treatment showed 62% (13/21) of treatment-naïve patients, 82% (9/11) of prior relapsers and 73% (22/30) of prior nonresponders achieved SVR12 (Table 2).
Virological response in patients, by immunosuppressant regimen and overall.
| n/N (%) | TAC (n = 50) | CsA (n = 24) | All (n = 74) |
|---|---|---|---|
| RVR | 19/50 (38) | 4/24 (17) | 23/74 (31) |
| eRVR | 19/50 (38) | 4/24 (17) | 23/74 (31) |
| SVR12 | |||
| All | 33/50 (66) | 20/24 (83) | 53/74 (72) |
| Treatment naïve | 4/11 (36) | 9/10 (90) | 13/21 (62) |
| Prior relapser | 7/9 (78) | 2/2 (100) | 9/11 (82) |
| Prior non-responder | 14/19 (74) | 8/11 (73) | 22/30 (73) |
| Genotype 1a* | 11/17 (65) | 8/11 (73) | 19/28 (68) |
| Genotype 1b* | 19/30 (63) | 11/12 (92) | 30/42 (71) |
| No or minimal fibrosis | 26/34 (77) | ||
| Portal fibrosis | 21/32 (66) | ||
| Bridging fibrosis | 6/8 (75) |
RVR: rapid virological response defined as HCV RNA < 25 IU/mL, undetected at week 4 of treatment. eRVR: extended rapid virologie response defined as HCV RNA < 25 IU/mL, undetected at week 4 and 12 of treatment. SVR12: sustained virological response defined as HCV RNA < 25 IU/mL 12 weeks after the last planned dose of study medication. * Two patients with HCV genotype 1d were also enrolled and both patients achieved SVR12.
In patients who had received a liver transplant 1-3 years prior to study enrollment, 32/43 (74%; 95% CI: 58.8 to 86.5) patients achieved SVR12, as did 19/25 (76%; 95% CI: 54.9 to 90.6) patients who had received a transplant > 3 years prior to the study start. Too few patients had received a liver transplant < 1 year prior to enrollment (n = 2) to allow meaningful comparison of SVR12 rates (Supplementary Figure 2).
SVR12 rates were similar in patients with HCV genotype 1b (71%, 95% CI: 55.4 to 84.3) and genotype 1a infection (68%, 95% CI: 47.6 to 84.1). In terms of IL28B status, the highest SVR12 rates were seen in patients with the CC genotype (94%, 95% CI: 69.8 to 99.8) compared with the CT and TT genotypes (68%, 95% CI: 50.9 to 81.4 and 61%, 95% CI: 35.7 to 82.7, respectively).
Overall, 31% (23/74) of patients had rapid virological response (RVR), defined as HCV RNA < 25 IU/mL, target not detected at week 4. Of the patients receiving TAC, 38% (19/50) achieved RVR, as did 17% (4/24) of those receiving CsA (Table 2). All patients who achieved RVR (23/74) went on to achieve extended RVR (eRVR), defined as HCV RNA < 25 IU/mL, target not detected at weeks 4 and 12 (Table 2).
VirologyOn-treatment virologic failure was observed in three (14%) treatment-naïve patients, one prior relapser (9%) and three (10%) prior non-responders. Five of the seven patients (71%), who experienced on-treatment virologic failure and also had sequencing data, had high-level telaprevir-resistant variants at the time of failure. One patient (14%) had lower-level telaprevir-resistant variants and one patient (14%) had wild-type virus.
All four patients with HCV genotype 1a virus who experienced on-treatment virologic failure had higher-level telaprevir-resistant variants V36M+R155K. Of the patients with HCV genotype 1b virus who experienced ontreatment virologic failure, one patient had a higher-level telaprevir-resistant variant (T54S+A156S), one patient had a lower-level telaprevir resistant variant (V36L+T54S/T), and one patient had wild-type virus.
In patients who relapsed, one patient was treatment naïve (5%) and three patients (10%) were prior non-responders. Overall, lower-level telaprevir-resistant variants were present in three of these four patients (75%) at the time of relapse, one patient had wild-type virus, and all four of these patients had HCV genotype 1b.
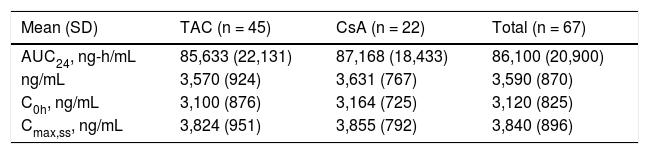
Pharmacokinetic analyses of TVRThe steady-state predose (C0h) and maximal (Cmax,ss) concentrations of TVR, and area under the curve 24 hours after dosing (AUC24) were comparable between the patients receiving TAC and CsA (Table 3). The mean steady-state concentrations of TVR (Css,avg) were similar between patients receiving TAC and CsA, and within the expected ranges when TVR is dosed q8h (Table 3).
Telaprevir concentrations and exposure in the study population.
| Mean (SD) | TAC (n = 45) | CsA (n = 22) | Total (n = 67) |
|---|---|---|---|
| AUC24, ng-h/mL | 85,633 (22,131) | 87,168 (18,433) | 86,100 (20,900) |
| ng/mL | 3,570 (924) | 3,631 (767) | 3,590 (870) |
| C0h, ng/mL | 3,100 (876) | 3,164 (725) | 3,120 (825) |
| Cmax,ss, ng/mL | 3,824 (951) | 3,855 (792) | 3,840 (896) |
AUC24: area under the plasma concentration-time curve over 24 h. Css,avg: average steady-state plasma concentration. C0h: trough plasma concentration. Cmax,ss: maximum plasma concentration at steady state.
At screening, the mean (SE) daily dose of TAC was 2.34 (0.222) mg, which was reduced to 1.87 (0.236) mg at baseline and further reduced on day 2 of treatment to 0.72 (0.217) mg. The lowest mean dose of TAC when coadministered with TVR was 0.21 (0.045) mg at week 3 (Figure 1A). Following the completion of the telaprevir treatment phase, mean daily doses of TAC ranged from 3.25 (0.275) mg/day to 3.56 (0.478) mg/day; in some patients, this TAC dose was higher than the initial pre-TVR dosing.
 tacrolimus and B) cyclosporin A and the mean plasma concentrations of C) tacrolimus and D) cyclosporin A during the study. Dashed lines denote the therapeutic range of concentrations.")
The mean dose of CsA at screening was 151.0 (11.00) mg, which was reduced to 86.3 (11.90) mg at baseline. Further reductions of the mean daily CsA dose to between 47.3 (3.96) mg and 57.6 (8.53) mg occurred during the TVR treatment phase (Figure 1B). Following the end of the TVR administration, mean daily doses of CsA were increased to between 150.2 (9.80) mg and 165.3 (7.13) mg, which were in the range of pre-TVR doses.
Therapeutic levels of TAC and CsA were largely maintained by the investigator during TVR treatment by adjusting the dose of TAC or CsA and prolonging the TAC dosing interval. Investigator-directed reductions in immunosuppressant dose made at baseline for all patients ensured maintenance of therapeutic concentrations of TAC and CsA throughout the TVR treatment period (Figure 1C and 1D).
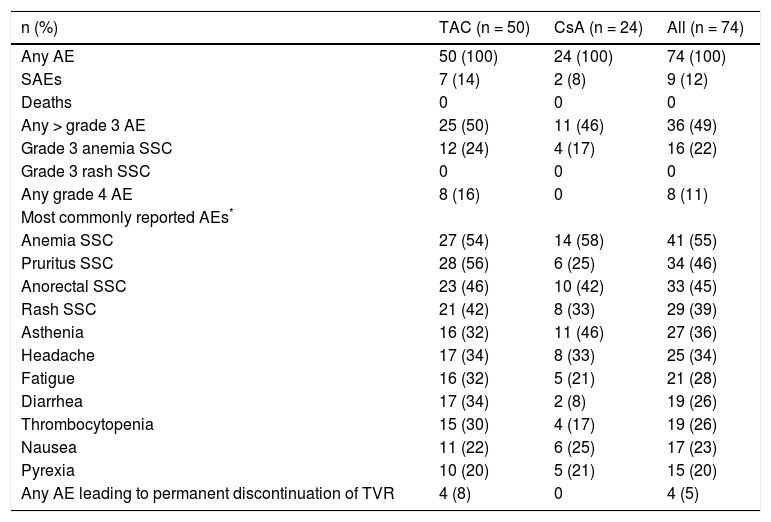
SafetyDiscontinuation of treatment with TVR due to AEs was infrequent with a rate of 5% (four patients: one case each of neutropenia and thrombocytopenia, abdominal pain, cholestasis, and urinary tract infection). Discontinuation of PR treatment due to AEs occurred in 12 (16%) patients.
Overall, AEs were reported by 100% of patients, with 12% (9/74) reporting serious AEs (SAEs). No patients died in the study. The most commonly reported AEs during the TVR treatment phase were anemia (55%, 41/74), pruritus (46%, 34/74), anorectal symptoms (45%, 33/74), and rash (39%, 29/74). AEs of grade 3 or greater were reported by 49% (36/74) of patients, and 11% (8/74) of patients experienced grade 4 AEs (Table 4).
Adverse events reported during the telaprevir treatment phase.
| n (%) | TAC (n = 50) | CsA (n = 24) | All (n = 74) |
|---|---|---|---|
| Any AE | 50 (100) | 24 (100) | 74 (100) |
| SAEs | 7 (14) | 2 (8) | 9 (12) |
| Deaths | 0 | 0 | 0 |
| Any > grade 3 AE | 25 (50) | 11 (46) | 36 (49) |
| Grade 3 anemia SSC | 12 (24) | 4 (17) | 16 (22) |
| Grade 3 rash SSC | 0 | 0 | 0 |
| Any grade 4 AE | 8 (16) | 0 | 8 (11) |
| Most commonly reported AEs* | |||
| Anemia SSC | 27 (54) | 14 (58) | 41 (55) |
| Pruritus SSC | 28 (56) | 6 (25) | 34 (46) |
| Anorectal SSC | 23 (46) | 10 (42) | 33 (45) |
| Rash SSC | 21 (42) | 8 (33) | 29 (39) |
| Asthenia | 16 (32) | 11 (46) | 27 (36) |
| Headache | 17 (34) | 8 (33) | 25 (34) |
| Fatigue | 16 (32) | 5 (21) | 21 (28) |
| Diarrhea | 17 (34) | 2 (8) | 19 (26) |
| Thrombocytopenia | 15 (30) | 4 (17) | 19 (26) |
| Nausea | 11 (22) | 6 (25) | 17 (23) |
| Pyrexia | 10 (20) | 5 (21) | 15 (20) |
| Any AE leading to permanent discontinuation of TVR | 4 (8) | 0 | 4 (5) |
AEs reported by ≥ 20% of patients overall. SSC: special search category. Anemia SSC: anemia, hemoglobin decreased, pancytopenia. Pruritus SSC: pruritus, pruritus generalized. Anorectal SSC: hemorrhoids, anal pruritus, or discomfort. Rash SCC: cheilitis, dermatitis, eczema, erythema, rash, rash erythematous, rash maculo-papular, or rash papular.
Of the patients who experienced anemia, 37% (27/74) had a reduction in RBV dose and 47% (35/74) required supportive treatment, with 42% (31/74) receiving an erythropoiesis-stimulating agent and 22% (16/74) receiving a blood transfusion.
Elevated serum creatinine levels occurred in 46% (34/74) of patients during the TVR treatment phase, with most being grade 1 (26%, 19/74) in severity (defined as ≥ 1.1 - ≤ 1.3 times the upper limit of normal) and occurring with a similar incidence between the TAC and CsA groups. Only one patient (1%) experienced an increase in creatinine that was considered grade 3 in severity (defined as > 1.8 - ≤ 3.4 times the upper limit of normal).
Graft rejection occurred in six patients (8%) during the study, three of whom were receiving CsA and three who were receiving TAC. Graft rejection did not occur during the TVR treatment phase, and none of these cases resulted in graft loss or were considered by investigators to be related to TVR. Of the patients receiving CsA, one patient with a grade 2 acute liver rejection at week 29 continued on the same PR dose, and two patients with a grade 3 event discontinued PR permanently at weeks 24 (liver transplant rejection) and 35 (suspected graft rejection), respectively. In the patients receiving TAC, one patient experiencing a grade 2 liver graft rejection at week 20 permanently discontinued PR. At week 24 and 25, two other patients experienced grade 3 and 4 liver graft rejection events, respectively, but continued treatment with no changes to PR dosing. Patients who received systemic corticosteroids to treat the rejection stopped PR treatment per protocol; for the patients continuing PR treatment, no dosage changes were made. All the patients recovered without liver graft loss.
Liver graft histologyThe majority of liver graft biopsies taken from patients 24 weeks after treatment completion (67%, 34/51) showed no change in METAVIR fibrosis stage; 18% (9/51) showed improvement and 16% showed progression of fibrosis (8/51). A similar trend was seen for METAVIR overall inflammatory activity, whereas Ishak-assessed inflammatory activity improved between pre- and posttreatment in the majority of patients, and in more patients receiving TAC compared with CsA. There was no difference in fibrosis seen between genotype 1a and 1b patients, but more patients with genotype 1a than 1b experienced an improvement in overall inflammatory activity (45% compared with 20%).
In terms of graft rejection, no biopsies showed definitive histological evidence of acute or chronic rejection or plasma cell hepatitis 24 weeks after the last actual dose of study drugs, although two specimens showed indeterminate or uncertain evidence of acute rejection.
DiscussionPatients who develop chronic HCV infection can progress to develop severe liver disease and may require a liver transplant. Following transplantation, recurrence of HCV infection is almost universal, and associated with an accelerated progression of graft fibrosis. Eradicating HCV infection in these patients is therefore a priority.
In this study, SVR12 rates of 72% (53/74, 95% CI: 59.9 to 81.5) were achieved in post-liver transplant patients without cirrhosis with recurrent HCV treated with TVR in combination with PR. This indicates improved efficacy over treatment with PR alone.11 The findings in our larger cohort are consistent with an initial small study treating recurrent HCV in post-transplant patients with TVRbased triple therapy that demonstrated improved rates of SVR over historical rates with PR, with five out of nine patients achieving SVR24.28 The SVR rates in patients with F0-1 and F2 fibrosis were relatively similar, but it should be noted that patients enrolled in the REPLACE study had less advanced fibrosis, with F3 fibrosis present in only 11% of patients, in contrast to other studies which enrolled a higher proportion of patients with advanced fibrosis and cirrhosis.32,33 Higher SVR12 rates were seen in the REPLACE study than in two other recently published studies evaluating virological response to TVR-based therapy in more ‘difficult-to-treat’ groups of post-liver transplant patients; those patients had more severe liver disease (42-53% had F3-F4 fibrosis) and, in some cases, had already received previous dual therapy for post-transplant recurrence of HCV infection.31,32
Overall, low numbers of patients experienced on-treatment virological failure (11%) or relapse (5%). These data and the resistance profile of patients who experienced relapse or on-treatment virologic failure are consistent with findings in non-transplant patients treated with triple therapy, where approximately 10% of patients experienced ontreatment virological failure and approximately 10% relapsed.12–14 The study size did not allow for investigation into predictive factors of SVR response in patients receiving TVR in combination with PR.
Safety findings in this study with TVR in combination with PR in liver transplant recipients were consistent with previous studies, which showed more safety and tolerability issues than in non-transplant patients;27,28,34,35 overall, 12% of patients reported SAEs, 49% (36/74) reported grade 3 or higher AEs and 11% experienced grade 4 AEs. However, only four patients (5%) permanently discontinued treatment with TVR due to an AE, which is similar to non-transplant patients.12–14
Consistent with previous findings that anemia is a common occurrence in liver transplant recipients treated for recurrent HCV infection with PR therapy alone,36 the REPLACE study demonstrated an increased rate of anemia in patients receiving TVR in combination with PR compared with non-transplant patients, despite using a lower RBV dose. However, the rates of measured hemoglobin decrease between transplant and non-transplant patients were similar.12–14 These data are also consistent with a previous study where 65% (23/35) of patients treated with TVR-based triple therapy, and 78% (32/41) of patients receiving BOC-based triple therapy developed anemia, and 63% of the total patient population received a reduced dose of RBV.30 Rates of creatinine elevation in the REPLACE study were consistent with the post-liver transplant population studied and the co-administered immunosuppressants. Renal dysfunction is not uncommon in post-transplant patients and calcineurin inhibitor nephrotoxicity is frequently the cause. However, it is not the only source of post-liver transplantation dysfunction. A review of 23 biopsies taken from 54 (out of a total of 2100) liver transplant patients from a single center from 1996 onwards demonstrated several different causes of renal dysfunction resulting in increased creatinine.37 The histological findings from these biopsies showed calcineurin inhibitor nephrotoxicity (48%), hypertensive changes (44%), membranoproliferative glomerulonephritis (17%), immunoglobulin A nephropathy (9%), diabetic nephropathy (9%), crescentic glomerulonephritis (4%) and acute tubular necrosis (4%).
As many DAAs are substrates and/or inhibitors of cytochrome P450 (CYP) 3A4 and P-glycoprotein, triple therapy of recurrent HCV is burdened by significant potential for drug-drug interactions with the immunosuppressants TAC and CsA.14, 17–23 Mean plasma concentrations of TAC and CsA are known to increase upon co-administration with TVR,27,28 yet increases in serum levels of immunosuppressant drugs have successfully been managed with dose modification of TAC and CsA.27 The REPLACE study demonstrated that, with adequate knowledge and information to support accurate anticipation of the extent of drug-drug interactions with TAC and CsA, investigators were able to successfully implement the necessary dose reductions. Additionally for TAC, decreased dose frequency at initiation of TVR treatment and incremental dose increases at the end of TVR treatment were also implemented. Accordingly, plasma concentrations were maintained at therapeutically active levels and no incidence of graft rejection or graft loss occurred during treatment with TVR. Overall, the rate of rejection in this study was similar to previously published studies using PR (5%).6 The effective use of TVR in this stable liver transplant patient population receiving TAC and CsA illustrates that it is possible to manage drug interactions between DAAs and immunosuppressants. These data may be enlightening in the design of future studies with DAAs in this population.
While data on the efficacy of second-generation treatments in a post-transplant setting is now becoming available,38–40 not all countries will have access to these treatments. As such, TVR may remain a useful therapeutic option in treating particular subsets of patients.
In summary, although drug-drug interactions impact dosing of CsA and TAC, these interactions can be managed clinically through modifications in TAC and CsA dose and TAC dosing intervals. TVR-based triple therapy in post-liver transplant patients with recurrent HCV infection is an effective therapy, with safety and tolerability profiles generally consistent with previous studies of TVR-based triple therapy in the post-liver transplant study population on calcineurin inhibitors. Rates of reported anemia were higher in this study than previous TVR-based triple therapy studies in non-transplant patients.
Abbreviations- •
AE: adverse event.
- •
AUC24: area under the curve 24 hours after dosing.
- •
AUCt: area under the curve over the dosing interval.
- •
Coh: initial concentration.
- •
CI: confidence interval.
- •
Cmax,ss: maximal steady-state concentration.
- •
CsA: cyclosporin A
- •
Css,avg: average steady-state plasma concentration.
- •
Ctrough: pre-dose plasma concentration.
- •
CYP: cytochrome P450.
- •
DAA: direct-acting antiviral.
- •
eRVR: extended RVR.
- •
HCV: hepatitis C virus.
- •
LLOQ: lower limit of quantification.
- •
Peg-IFN/P: pegylated interferon.
- •
q8h: every 8 hours.
- •
RBV/R: ribavirin.
- •
RVR: rapid virological response.
- •
SAE: serious AE.
- •
SD: standard deviation.
- •
SE: standard error.
- •
SVR: sustained virologic response.
- •
TAC: tacrolimus.
- •
TVR: telaprevir.
X Forns has received unrestricted grant support from Janssen Pharmaceuticals, and acted as an advisor for AbbVie, Janssen Pharmaceuticals, and Gilead Sciences; D Samuel has been a consultant for AbbVie, Astellas, Gilead Sciences, Janssen Pharmaceuticals, Merck Sharp & Dohme, and Novartis; D Mutimer has been a speaker/consultant to Janssen Pharmaceuticals and other pharmaceutical companies; S Fagiuoli has acted as consultant/speaker for Abbvie, Astellas, Bayer, Biotest Pharmaceuticals, Bristol-Myers Squibb, Gilead Sciences, Grifols, Kedrion, Janssen Pharmaceuticals, Merck Sharp & Dohme, Novartis, and Roche; M Navasa has received unrestricted grant support from Novartis and has acted as an advisor for Novartis; K Agarwal has received grant support from Astellas, Gilead Sciences, and Roche and been a consultant/speaker for AbbVie, Astellas, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen Pharmaceuticals, Merck Sharp & Dohme, Novartis, and Roche; M Berenguer has been a speaker/consultant for AbbVie, Janssen Pharmaceuticals, Merck Sharp & Dohme, Novartis, and Roche; M Colombo has been a speaker/consultant for AbbVie, Alfa Wassermann, Bristol-Myers Squibb, Bayer, Boehringer Ingelheim, Gilead Sciences, GlaxoSmithKline, Janssen Pharmaceuticals, Jennerex Biotherapeutics Incorporated, Merck, Novartis, Roche, and Vertex, and has received grant/ research support from Bristol-Myers Squibb, Gilead Sciences, and Roche; K Herzer has received grant support from Astellas, Biotest, and Novartis, and been a consultant/ speaker for AbbVie, Biotest, Bristol-Myers Squibb, Gilead Sciences, Janssen Pharmaceuticals, Novartis, and Roche; F Nevens has received grant support from Astellas, Ipsen, Janssen Pharmaceuticals, Merck Sharp & Dohme, and Roche; B Daems, K Janssen, H Kimko, E Lathouwers, S Ouwerkerk-Mahadevan, R Van Solingen-Ristea, and J Witek are employees of Janssen Pharmaceuticals and may be Johnson & Johnson stockholders.
Financial SupportThis study was sponsored by Janssen Pharmaceuticals. Writing assistance was provided by Stephanie Gibson (Medical Writer) of Zoetic Science, an Ashfield company, part of UDG Healthcare plc, Macclesfield, UK; this support was funded by Janssen Pharmaceuticals.
The REPLACE study was registered with ClinicalTrials.gov (NCT01571583).
Authors’ ContributionsXavier Forns, Didier Samuel, David Mutimer, Kosh Agarwal, James Witek and Rodica Van Solingen-Ristea contributed to the conception and design of this study. Xavier Forns, Didier Samuel, David Mutimer, Stefano Fagiuoli, Miquel Navasa, Kosh Agarwal, Marina Berenguer, Massimo Colombo, Kerstin Herzer and Frederik Nevens recruited patients into the study and participated in data collection. All authors contributed to data analysis. All authors critically reviewed the article for important intellectual content and approved the final content for submission.
AcknowledgementsWe express our gratitude to the patients and their families, the REPLACE study investigators including: Pietro Andreone, Rafael Barcena Marugan, Susanne Beckbaum, Luca Belli, Patrizia Burra, José Luis Calleja, Luis Castells, Maria Francesca Donato, Francois Durand, Peter Ferenci, Guido Gerken, Dominique Guyader, Marianne Latournerie, Andreas Maieron, Zoe Marino, Christophe Moreno, George-Philippe Pageaux and Heiner Wedemeyer, Andrew Bathgate for his role in the Safety Monitoring Committee, and Stefan G. Hübscher for analysing the patient biopsy samples.
 patient disposition.")




