





Hepatic encephalopathy (HE) is a brain dysfunction caused by liver insufficiency and/or portosystemic shunts. Between 30%–40% of patients with cirrhosis will present overt HE during their lifetime. While the pathophysiology of HE is not entirely understood, three critical factors have been identified: hyperammonaemia, systemic inflammation and oxidative stress by glutaminase gene alterations. Minimal HE is defined by the presence of signs of cognitive abnormalities in a patient without asterixis or disorientation; it can only be diagnosed with neuropsychological or psychometric tests. The diagnosis of overt HE is based on clinical examination with clinical scales. Currently, only overt HE should be routinely treated. The aims of treatment in an acute episode should be to improve the mental status, identify and treat the precipitating factor, reduce duration and limit consequences. Treatment strategies are targeted at reducing ammonia production and/or increasing its elimination. Even though minimal HE has negative effects on the patient's quality of life and effects on prognosis, indications for treatment are still controversial. There are still many unanswered questions regarding the pathophysiology and management of HE. We should also endeavor to develop more accurate and objective diagnostic methods for overt HE that would permit early detection and help improve outcomes on quality of life and economic burden.
Hepatic encephalopathy (HE) is a brain dysfunction caused by liver insufficiency and/or portosystemic shunts. It has a broad spectrum of neuro-psychiatric manifestations ranging in severity from subclinical alterations to coma [1]. In cirrhosis, the appearance of overt HE is a sign of decompensation and portends stage IV disease (the first non-bleeding decompensation) [2]. The cumulative incidence of HE in cirrhosis is 5–25% at five years and 7–42% at 10 years. The prevalence of covert HE is 20–80% and 30–40% of patients with cirrhosis will present overt HE during their lifetime [3]. Despite appropriate treatment, patients with a previous history of overt HE have a 42% risk of recurrence within one year [4]. The economic burden is important; a systematic review showed that annual HE direct costs range from $5320 to $50,120 US [5]. In this review, we will be focusing on HE associated with cirrhosis.
2PathophysiologyThe pathophysiology of HE is not entirely understood; however, three critical factors have been identified: hyperammonaemia, systemic inflammation and oxidative stress by glutaminase gene alterations (Fig. 1) [6].
 is located in the intestines, liver and brain. PAG catalyses the hydrolysis of glutamine into ammonia in patients with cirrhosis; intestinal concentrations of PAG are higher. It has also been found that long alleles in the microsatellites in the PAG gene are associated with increased risk of HE.")
Physiopathology of hepatic encephalopathy.
1 Ammonia is primarily produced in the gut by protein digestion and bacterial urease activity; changes in microbiome have also been associated with increased endotoxin levels. 2 Alterations in the intestinal barrier cause bacterial translocation which leads to cytokine production, resulting in systemic inflammation. 3 The liver regulates ammonia concentration through the urea cycle, transforming ammonia into urea, which is then excreted by the kidneys in urine; when its capacity is exceeded, hyperammonaemia occurs. 4 Ammonia can cross the blood brain barrier, where the only cells that are able to metabolize it are astrocytes using glutamine synthetase, which transforms ammonia into glutamine. Glutamine acts as an osmolyte causing neuronal and astrocyte swelling leading to brain oedema and alterations in brain metabolism. 5 Phosphate-activated glutaminase (PAG) is located in the intestines, liver and brain. PAG catalyses the hydrolysis of glutamine into ammonia in patients with cirrhosis; intestinal concentrations of PAG are higher. It has also been found that long alleles in the microsatellites in the PAG gene are associated with increased risk of HE.
Ammonia is the product of protein digestion, amino acid deamination and bacterial urease activity. It is mostly produced in the gut and carried into the liver by the portal vein. Since high concentrations of ammonia are toxic, the liver regulates the concentration in systemic circulation through the urea cycle, maintaining low levels (30–50 µM) [4, 7].
The skeletal muscle also contributes to the regulation of blood ammonia levels by absorbing ammonia from plasma and releasing glutamine. In patients with decompensated cirrhosis, the muscle uptake of branched chain amino acids increases with the arterial ammonia concentration. Hence, hyperammonaemia lowers amino acid plasma levels by increasing muscular metabolism and glutamine synthesis [8]. In cirrhosis, the muscle may remove even more ammonia than the liver [8].
Ammonia can cross the blood brain barrier, where the only cells that metabolize ammonia are astrocytes by means of the enzyme glutamine synthetase, which transforms ammonia into glutamine. Glutamine acts as an osmolyte and its accumulation leads to astrocyte swelling, followed by brain oedema and metabolic dysfunction. [6, 7]. It can also directly affect cell function by changing the pH, causing oxidative stress and mitochondrial dysfunction, raising the membrane potential of both neurons and astrocytes, and altering brain metabolism [9].
2.2InflammationPatients with cirrhosis have an impaired capacity to clear bacterial antigens arriving from the gut. Furthermore, damage to the intestinal barrier results in a higher rate of intestinal bacteria translocation, producing pathogen-associated molecular patterns (PAMPs), which interact with Kupffer cells leading to activation of the immune response and systemic inflammation [4, 6, 10, 11].
It has been found that hyperammonaemia is associated with significant neuropsychological deterioration during the inflammatory state, secondary to infection in patients with cirrhosis [12]. Another prospective study found that the Systemic Inflammatory Response Syndrome score was higher in patients admitted for grade 4 HE, suggesting an association between inflammation and HE [13].
Microbiota also play a critical role since the function and composition of microbiota may alter the disease's course [4]. It has been found that overt HE is associated with alterations in the stool microbiome compared with non-cirrhotic patients. HE is also strongly associated with cognitive impairment [14]. Changes in microbiome become more severe with decompensation—the higher the endotoxin levels, the lower the dysbiosis rate [15]. Also, when controls were compared with patients with HE, a significant reduction in autochthonous taxa, such as Clostridiales XIV, Ruminococcaceae and Lachnospiraceae was found, with a significant increase in pathogenic taxa, for example, Enterococcaeae, Staphylococcaceae and Enterobacteriaceae[15].
2.3Glutaminase genePhosphate-activated glutaminase catalyzes the hydrolysis of glutamine into glutamate and ammonia; it is found in the intestine, kidney and neurons [716] Intestinal phosphate-activated glutaminase concentrations are higher in patients with cirrhosis compared with healthy subjects; glutaminase concentrations are also independently associated with the presence of minimal HE [17].
Having two long alleles of a microsatellite in the phosphate-activated glutaminase gene (kidney type) correlates with higher glutaminase concentrations in vivo and an increased risk for overt HE [18]. Another study found that carriers of the long microsatellite had a higher risk of HE diagnosed by critical flicker frequency test (Odds Ratio (OR) 3.23, 95% confidence interval (CI) 1.46–7.13; P = 0.004) [19].
3DiagnosisHE can be classified based on four different factors: (1) underlying cause, (2) severity of disease manifestation (according to West-Haven Criteria, WHC and International Society of Hepatic Encephalopathy and Nitrogen Metabolism, ISHEN criteria), (3) time course and (4) precipitating factors (Table 1) [1, 3].
Minimal and covert HE is very difficult to diagnose based only on clinical examination. They are defined by the presence of signs of cognitive abnormalities in a patient without asterixis or disorientation. Minimal HE indicates that there are no clinical signs; therefore, it can only be diagnosed with neuropsychological or psychometric tests. On the other hand, covert HE is defined as grade I in the WHC (Table 1) [1, 20, 21].
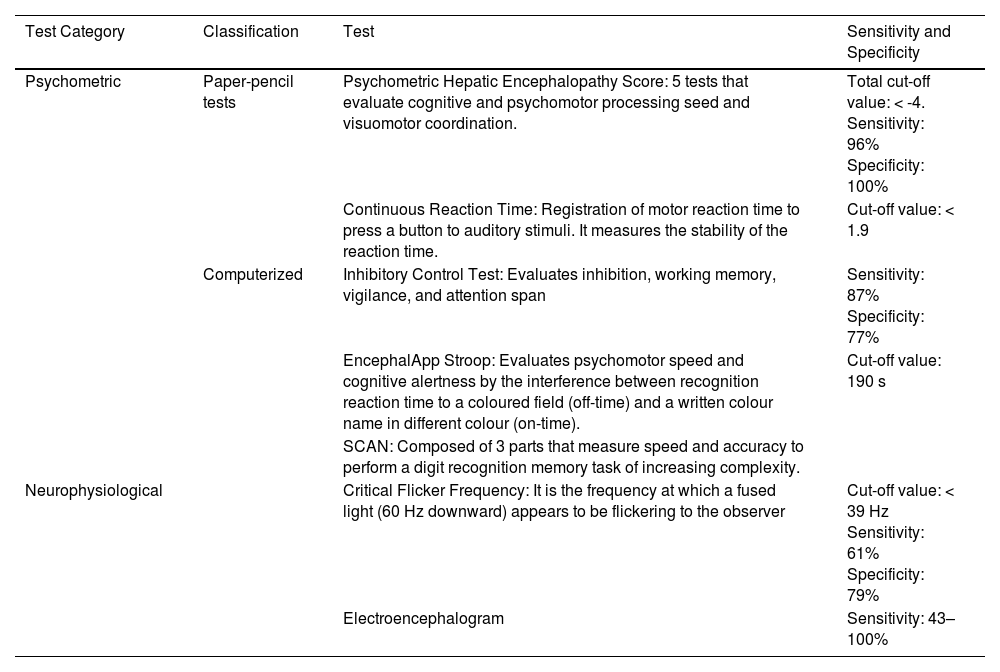
Testing strategies can be divided into two categories: psychometric and neurophysiological (Table 2). The gold standard of diagnosis is the Psychometric Hepatic Encephalopathy Score. [1, 20]. By consensus, the diagnosis of minimal HE or covert HE should be made using two testing strategies: the Psychometric Hepatic Encephalopathy Score and one of a range of computerized and validated neurophysiological tests. It should be noted that none of the tests is specific for HE [22].
The diagnosis of overt HE is largely based on clinical examination. Clinical scales are used for diagnosis and grading severity, but they are subjective and require clinical skills. [1, 6, 23]. The gold standard of diagnosis according to the European Association for the Study of the Liver/ American Association for the Study of Liver Diseases (EASL/AASLD) Guidelines is the WHC (Table 1), overt HE consists of stages 2–4 [1].
Nevertheless, neither the clinical symptoms nor the laboratory or imaging findings are specific to HE. For this reason, overt HE remains a diagnosis of exclusion and must always be differentiated from other neurologic diseases, other forms of metabolic encephalopathy, mental status alterations due to alcohol or drug consumption, medications, or effects of hyponatremia [20]. A conscientious clinical history should be taken in order to identify precipitating factors and prompt correction should follow. Previous HE episodes increase the likelihood of the current episode being HE and should be taken into consideration [6].
3.3Laboratory testingTo date, there are no laboratory markers able to diagnose overt HE, but they are useful to identify precipitating factors or to rule out other diagnoses [20]. While measuring ammonia levels might seem attractive due to its importance in pathogenesis, its role in diagnosis is still debated. One must remember that ammonia impacts cerebral function only after it crosses the blood brain barrier. For this reason, serum ammonia levels do not reflect the rate of ammonia entry into the brain [20]. However, if ammonia levels are normal in a patient suspected of overt HE, the diagnosis should be challenged [1].
The best evidence for the relationship between ammonia levels and HE severity is in the context of acute liver failure. In one study, a cut-off value of > 100 µmol/L identified patients at high risk of developing HE, and the authors also found that ammonia levels remain high in patients with intracranial hypertension [24]. Another study in hospitalized patients found that a cut-off value of 80 µmol/L was associated with higher 28-day mortality [25].
3.4Imaging testingCurrently, there is no image modality that can contribute to the diagnosis or grading of overt HE [4]. Magnetic resonance imaging is the most commonly used imaging method in research studies, and specific software can demonstrate structural and functional cellular changes [4]. Bilateral symmetric hyperintensity of the basal ganglia has been associated with excess manganese deposits; however, its relation with HE has not been demonstrated [26]. Diffusion-weight magnetic resonance has demonstrated interstitial oedema in patients with minimal HE or overt HE [27]. Positron emission tomography using 18F-fluorodeoxyglucose has demonstrated a reduction in glucose metabolism in the anterior cingulate gyrus that may be associated with attention deficit in patients with HE [28].
Brain imaging should only be performed if there are signs of other neurological issues or if there is an inadequate response to overt HE treatment [6]. It has been suggested that response to treatment might be the best measure to prove the diagnosis [23].
3.5ElectroencephalogramHE is associated with changes in two of the principal components of the electroencephalographic pattern: (1) the rhythmic background activity, which in patients with HE progressively slows down with the reduction of eye-opening reactivity and (2) transients. In patients with moderate to severe HE, "triphasic waves" can be observed in wake electroencephalogram (EEG). They can also be observed in other metabolic encephalopathies, where their appearance may be associated with white matter oedema [29].
A study found that EEG alterations have a prognostic value by predicting episodes of overt HE (HR 3.3, 95% CI 1.8–6.1) and mortality (HR 3.1, 95% CI 1.7–5.6) within one year [30]. A large study testing the benefit of adding an EEG-based HE index to the Model for End-Stage Liver Disease (MELD) score, found that it improved the accuracy to predict 12 month (AUCMELD-EEG=0.69 ± 0.03 vs. AUCMELD = 0.62 ± 0.04, P = 0.016) and 18 month (AUCMELD-EEG = 0.71 ± 0.03 vs. AUCMELD = 0.64 ± 0.03, P = 0.018) mortality [31].
Decreased variability or increased regularity of physiological rhythms has also been found in the EEG of patients with cirrhosis compared with the unimpaired population (21.2% vs. 22.4%, P < 0.001) [32]. However, EEG is nonspecific and may be influenced by many metabolic disturbances and alcohol and drug intake.
4ManagementHE is most commonly precipitated in patients with cirrhosis. The most common precipitating factors include infections, followed by a previous history of overt HE, sarcopenia, hyponatremia, type 2 diabetes mellitus, renal impairment, low albumin levels and medications (diuretics, proton pump inhibitors, beta-blocker [33]. and statins) [3].
The EASL/AASLD guidelines recommend that only overt HE should be routinely treated [1]. The aims of treatment in an acute episode should be to improve the mental status, identify and treat the precipitating factor, reduce duration and limit consequences. After one episode of overt HE, therapy should focus on preventing recurrence and hospital admissions, improving quality of life and limiting the impact on patients' families. [4, 20]. Treatment strategies are targeted at reducing ammonia production and/or increasing its elimination [34]. In order to prevent future events, attention should be focused on limiting and promptly correcting precipitating factors.
4.1Nonabsorbable disaccharidesThe disaccharides, lactulose and lactitol, are nondigestible substances and are the recommended first-line therapy. When they reach the colon, they are degraded into short-chain organic acids. The acidic environment reduces ammoniagenic bacteria, inhibits bacterial ammonia production and reduces ammonia absorption by converting ammonia to nonabsorbable ammonium. Furthermore, the increased osmolality also accelerates intestinal transit, removing excess faecal nitrogen through a laxative effect [4, 6, 20, 34]. Lactose also increases zinc absorption [35]. There is limited information from randomized control trials (RCT) supporting the use of this treatment.
A trial comparing lactulose enemas versus neomycin tablets found that both treatments have comparable effects in the improvement of asterixis and electroencephalograms, but lactose enemas also acidified stool pH and improved number-connection tests [36]. A trial comparing lactulose with lactitol found no significant difference in safety and efficacy [37]. Another RCT comparing acidifying enemas (lactitol and lactulose) improved HE compared with non-acidifying agents [38].
A systematic review of nonabsorbable disaccharides for the treatment of overt HE concluded that there is insufficient evidence to support or refute their use and should not serve as comparators in randomized trials, there was no effect on mortality (RR 0.41, 95 CI 0.42–2.04) [39]. A more recent Cochrane systematic review found that there is moderate quality evidence that disaccharides benefit outcomes mortality (RR 0.63, 95% CI 0.41–0.97) and HE (RR 0.58, 95% CI 0.50–0.69) when compared with placebo/no intervention, with a number necessary to treat (NNT): 6 [40].
Polyethylene glycol 3350-electrolyte (PEG) solution, an osmotic laxative, was compared with lactulose in the treatment of overt HE in hospitalized patients. Ninety-one percent of patients treated with PEG improved in one or more HE algorithm scores, compared with 52% in the control group (P < 0.01). The median time of HE resolution was two days with standard therapy and one day with PEG (P = 0.01) [41]. These findings suggest that PEG could be an alternative to lactulose in hospitalized patients.
4.2AntibioticsAntibiotics are aimed at reducing the number of ammonia producing bacteria in the intestine [34]. The first trial using antibiotics was performed with neomycin and demonstrated a reduction of ammonia colonic concentrations after cleansing with the antibiotic [42].
Rifaximin is a nonabsorbable antibiotic that acts against gram positive, gram negative, aerobic and anaerobic enteric bacteria by inhibiting RNA synthesis [4]. One of the first trials compared rifaximin with neomycin, and found that 1200 mg of rifaximin a day was as effective as neomycin in reducing HE grade at 30 days [43]. When compared with lactulose, rifaximin was found to be as safe and effective for HE reversal (95.4% and 84.4%, respectively, P = 0.315) [44]. The same was found when compared with lactitol (81.6% for rifaximin and 80.4% for lactitol) [45]. However, a randomized trial found that the combination of lactulose plus rifaximin is more effective than lactulose alone for HE reversal (76% and 50.8%, respectively, P < 0.004) and mortality reduction (23.8% vs. 49.1%, P < 0.05) [46].
A systematic review found that rifaximin is as effective as nonabsorbable disaccharides and other antibiotics (OR 0.96, 95% CI 0.94–4.08) but with a better safety profile for improving the mental status and reducing asterixis (OR 0.27, 95% CI 0.12–0.59) [47]. Rifaximin has also been associated with the reduction in healthcare costs by reducing hospitalization risk by 50% compared with lactulose [5]. A 6-month treatment with rifaximin maintains remission more effectively than placebo by reducing the risk of an episode of HE (HR 0.42, 95% CI 0.28–0.64; P < 0.001) and reducing the risk of hospitalization (HR 0.50, 95% CI 0.29–0.87; P = 0.01) [48].
4.3Metabolic ammonia scavengersMetabolic ammonia scavengers promote ammonia excretion through the urine by alternate pathways. Sodium benzoate, which conjugates with glycine in the liver and is excreted in the urine as hippurate, was one of the first proposed scavengers [49]. It was later tested and shown to be as efficacious as nonabsorbable disaccharides for the treatment of HE [50].
Glycerol phenylbutyrate also provides a surrogate pathway for ammonia removal in the form of urinary excretion of phenylacetyl glutamine. When compared with lactulose and rifaximin, it reduced total events (35% vs. 57%; P < 0.04) and time to first event (HR 0.56, 95% CI: 0.32–0.99; P < 0.05,) and ammonia levels were significantly lower (62 vs. 76 mmol/L; P < 0.04) [51].
Ornithine phenylacetate enhances the activity of glutamine synthetase, inducing muscle to trap ammonia as glutamine, which is then conjugated with phenylacetic acid to form phenylacetylglutamine and excreted in the urine [52]. In a trial, patients given ornithine phenylacetate after an episode of upper gastrointestinal bleeding showed that it reduced plasma ammonia levels (37% at day 5) and was safe [53]. A more recent trial found that ammonia levels did not differ when comparing ornithine phenylacetate with placebo. However, clinical improvements occurred 21 hours sooner in patients receiving ornithine phenylacetate and the safety profile was similar [52]. More clinical trials are warranted to make recommendations on these agents.
4.4L-ornithine-L-aspartateL-ornithine-L-aspartate reduces blood ammonia levels by providing substrate to stimulate the urea and the glutamine cycles, resulting in the conversion of ammonia into urea and glutamine. [20, 34]. It was demonstrated in a RCT that intravenous L-ornithine-L-aspartate improves mental state and lowers ammonia levels when compared with placebo [54]. Another study compared oral administration of L-ornithine-L-aspartate with lactulose and found no difference in the efficacy for improvement in mental status, asterixis, and decrease in ammonia level [55].
A recent Cochrane systematic review found that there are possible beneficial effects of L-ornithine-L-aspartate on mortality in comparison with placebo or no-intervention (Relative Risk, (RR) 0.42, 95% CI 0.24–0.72; I2 = 0%), and HE improvement (RR 0.70, 95% CI 0.59–0.83; I2 = 62%) with a NNT of 6.7. Nevertheless, the quality of evidence is very low and recommendations cannot be made [56].
4.5AlbuminAlbumin is a protein synthesized in the liver that has been used for many years as a volume expander. However, it has been found that albumin has other properties, such as modulating anti-inflammatory response and detoxification. [4, 6].
A trial that randomized patients to receive standard treatment (lactulose and rifaximin 1200 mg/day) with either 1.5 g/kg on day one and 1 kg on day three of albumin or isotonic saline found that there was no improvement in resolution of HE (57.7% vs. 53.5%, respectively; P > 0.05), notwithstanding, a difference in survival at day 90 (69.2% vs. 40.05, respectively; P = 0.02), suggesting that this group of patients could benefit from albumin infusion during hospitalization [57]. Subsequently, a trial that compared the use of lactulose alone versus lactulose plus albumin found that the addition of albumin was more efficient in reversing HE (75% vs. 53.3%; P = 0.03) and reducing mortality (18% vs. 31.6%; P < 0.05) [58].
4.6ProbioticsProbiotics are defined as living organisms that offer benefits to the host when administered in sufficient amounts by modulating gastrointestinal immunity [59]. It has been suggested that some probiotics may reduce inflammation, thus helping to restore the microbiota and preventing bacterial translocation by maintaining intestinal barrier integrity [60].
A recent meta-analysis comparing probiotics with lactulose showed the effects of probiotics on all-cause mortality (RR 5, 95% CI 0.25–102), adverse events considering the development of overt HE (RR 1.17, 95% CI 0.63 to 2.17), hospitalization (RR 0.33, 95% CI 0.04 to 3.07) and plasma ammonia concentration (MD 2.93 μmol/L, 95% CI 9.36 to 3.50) with very low-quality evidence. The most commonly used probiotic was VSL#3 [61].
The use of probiotics in minimal HE has rendered promising results. It has been demonstrated that the use of fiber with probiotics can reverse minimal HE [62]. A meta-analysis comparing probiotics with lactulose showed a reduction of risk of no improvement of MHE (RR 0.34, 95% CI 0.24–0.47; P < 0.0001) [63].
4.7Dietary managementThe degree of decompensation, portal-systemic shunting and nutritional status are important in each patient's response to diet. It is important to note that although proteins can precipitate HE, deprivation of protein leads to depletion of hepatic protein stores and malnutrition. For this reason, a small trial was performed testing the effects of vegetal protein and psyllium plantago compared with animal protein in patients with HE and type 2 Diabetes Mellitus. No difference was found in the incidence [64].
The current recommendations from the International Society for Hepatic Encephalopathy and Nitrogen Metabolism Consensus suggest a daily intake of 35–40 kcal/kg body weight and 1.2–1.5 g/kg protein daily, regardless of the presence of HE. Dairy and vegetable protein are also better tolerated [65].
4.8Occlusion of porto-systemic shuntsIn patients with persistent HE, spontaneous portocaval shunts should be suspected, particularly when liver function is not severely compromised. Clinical data is very limited in this group of patients. A retrospective analysis showed that this cohort of patients could benefit, with a decreased number of HE episodes at 100 days post embolization compared with before embolization (59.4% vs. 100%; P < 0.001), especially if their MELD score was < 11 [66].
Another retrospective study found that embolization of shunts was associated with improved survival in patients with MELD < 15 and no hepatocellular carcinoma when compared with no embolization (100% vs. 60%, respectively; P = 0.03) [67]. No RCT data is available at the moment, while other data suggest that it may be useful in a selection of patients with HE and, in most cases, should be considered a bridge to transplantation [4].
4.9Faecal microbiota transplantationAs has been previously noted, microbiota play a preeminent role in the pathogenesis of HE. An open-label RCT was conducted to verify the safety of faecal microbiota transplantation when compared with the standard of treatment in patients with recurrent HE. Patients undergoing standard of treatment had more serious adverse events than patients who underwent faecal microbiota transplantation (80% vs. 20%; P = 0.02). As a secondary outcome, cognition was also improved compared with baseline [68].
A 12-month follow-up after faecal microbiota transplantation corroborated its long-term safety. As a secondary outcome, there was sustained improvement in neurological status and a reduction in hospitalizations [69]. A systematic review found that faecal microbiota transplantation improved neurocognitive tests, and readmission rates were lower when compared with the standard of treatment. However, the quality of evidence is low and more RCTs are needed to make recommendations [70].
4.10ZincLow levels of zinc can increase blood ammonia levels by impairing both glutamine synthetase and the enzymes that participate in the urea cycle. A systematic review and meta-analysis showed that zinc supplementation improves performance in the number connection test (SMD -0.62; 95% CI –1.12 to -0.11) but does not reduce HE recurrence (RR 0.64, 95% CI 0.26–1.59) [71]. The only recommendation that can be made at this moment is to supplement patients who are zinc deficient.
4.11Secondary prophylaxisThe best quality evidence for secondary prophylaxis after an event of overt HE exists for lactulose and rifaximin. An open-label RCT showed that lactulose is effective in preventing the recurrence of overt HE at 14 months compared with placebo (19.6% vs. 46.8%; P < 0.001) [72]. Another trial found that it was also effective in preventing overt HE after acute variceal bleeding (14% vs. 40%; P = 0.03) [73]. Rifaximin has also been proposed as a long-term treatment. A trial showed that 550 mg twice daily of rifaximin for > 24 months reduces the rate of hospitalization when compared with placebo (0.21 vs. 0.72 events/PYE) [74].
Long-term albumin administration has also been proposed as a strategy to prevent overt HE recurrence. A recent RCT found, as a secondary outcome, that administration of 40 g of albumin twice weekly for two weeks and then 40 g weekly over 18 months reduced the incidence of grade 3–4 HE (Incidence rate ratio 0.48, 95% CI 0.37–0.63; P < 0.001) [75].
4.12Minimal hepatic encephalopathyEven though minimal HE has negative effects on patients's quality of life and affects prognosis, indications for treatment in minimal HE are still debated [76]. One small study in Mexico found that the prevalence of minimal HE in patients with decompensated cirrhosis to be up to 44% [77]. Therapy should be tailored to prevent progression to overt HE, reduce hospitalizations and improve quality of life [20].
A RCT comparing lactulose to no treatment in patients with minimal HE found that lactulose improved cognitive function with a significant decrease in abnormal neuropsychological tests from baseline to 3 months (2.74 to 0.75; P = 0.001) and quality of life [78]. Another study found that treatment with rifaximin over an 8-week period improved driving stimulator performance compared with placebo; 91% improved their cognitive performance compared with 61% of patients given placebo (P < 0.01) [79].
A more recent study found that rifaximin also improves the quality of life and cognitive function in patients with minimal HE, with a significant decrease in abnormal neuropsychological tests from baseline to 8 weeks (2.35 to 0.81; P = 0.000) [80]. A meta-analysis showed that the use of prebiotics, probiotics and symbiotics (RR 0.49, 95% CI 0.32–0.50; P < 0.001) and lactulose (RR 0.34, 95% CI 0.24–0.47; P < 0.0001) for the treatment of minimal HE is associated with a decrease in the risk of no improvement of minimal HE [81]. An open-label trial showed that treatment with lactulose, probiotics or a combination of both was equally effective in the improvement of minimal HE in 51–56% of patients [82].
A RCT found that a 60-day oral course of L-ornithine-L-aspartate was no better than placebo to improve minimal HE, but it did prevent future episodes of overt HE at 6 months (5% vs. 37.9%; P = 0.016) [83]. A systematic research and network meta-analysis found that rifaximin (OR 7.53; 95% PrI 4.45–12.73; SUCRA 89.2%) and lactulose (OR 5.39; 95% PrI 3.60–8.0; SUCRA 67.2%) are the most effective treatments for the reversal of minimal HE; L-ornithine-L-aspartate (OR 0.19; 95% PrI, 0.04–0.91; SUCRA 75.1%) and lactulose (OR 0.22; 95% PrI, 0.09–0.52; SUCRA 73.9%) are the most effective in preventing overt HE; and lactulose was the only agent that improved quality of life (2 RCTs; MD –6.34; 95% CI, –7.58 to –5.10) [84].
It is known that patients with minimal HE have impaired driving skills and an increased risk of motor vehicle accidents. In a cost-effectiveness trial, lactulose was the most cost-effective approach and rifaximin was not cost saving at current prices [85].
5Quality of lifeQuality of life is further impaired in patients with cirrhosis and HE. Many patients with cirrhosis have insomnia, delayed sleep timing and excessive daytime sleepiness. However, any cause-effect relationship with HE is yet to be defined [86]. Cirrhosis has also been associated with higher in-hospital mortality after motor vehicle accidents (OR: 3.5, 95% CI 2.5–5.5) [87].
A prospective study also found that minimal HE is associated with falls when compared with patients with cirrhosis without minimal HE (40.4% vs. 6.2%; P < 0.001). Cognitive dysfunction was the only independent predictive factor of falls (OR 10.2, 95% CI 3.4–30.4; P < 0.001) [88]. Treatment of minimal HE with lactulose has been found to improve quality of life compared with patients with no therapy (12.63 ± 15.10 vs. 6.36 ± 12.16, P = 0.0523) [89].
6Future perspectivesThe burden of HE on patients' quality of life, prognosis, mortality and healthcare costs is not negligible. Further efforts should be made to better understand the pathophysiology, impact of ammonia, the microbiota and systemic inflammation to help develop novel therapies with a direct aim.
We should also endeavor to develop more accurate and objective diagnostic methods for overt HE that would permit early detection and help improve outcomes on quality of life and economic burden. Minimal HE and covert HE should be defined more precisely as a way of facilitating clinical and treatment decisions. Cost-effectiveness studies should be made to delineate who should be treated and when therapy should be started and/or suspended.
While improving the mental status and shortening duration are the primary aims of treatment in acute bouts, focusing on the prevention of further episodes should be paramount for clinicians. There are still many questions unanswered for the correct diagnosis and management of HE and there is still a long way ahead of us to finally improve outcomes for these patients, their families and caregivers.
Author ContributionsPaulina Vidal-Cevallos: Conceptualization, data curation, visualization, writing, original draft. Norberto C. Chávez-Tapia: Conceptualization, data curation, funding acquisition, supervision, writing, review & editing. Misael Uribe: Conceptualization, data curation, funding acquisition, supervision, review & editing.



