





The clotting process is a dynamic array of multiple processes which can be described in four phases: platelet plug initiation and formation, clotting process propagation by the coagulation cascade, clotting termination by antithrombotic mechanisms and clot removal by fibrinolysis. The liver plays a central role in each of these phases of clotting process, as it synthesizes the majority of coagulation factors and proteins involved in fibrinolysis as well as thrombopoeitin, which is responsible for platelet production from megakaryocytes. Many pathological processes associated with cirrhosis, such as portal hypertension and endothelial dysfunction, as well as co-morbid conditions, may also alter the coagulation process. Consequently, patients with liver disease have a disturbed balance of procoagulant and anti-coagulant factors which deviates from the normal coagulation cascade. This situation poses an additional problem in the diagnostic and therapeutic approach to this group of patients, since traditional coagulation test may not be reliable for assessing bleeding or thrombotic risk and traditional transfusional strategies may not be applicable in cirrhotic patients. In this article, we review the pathophysiological bases of coagulation abnormalities, in cirrhotic patients, the diagnostic therapeutic strategies to be followed and its impact on the clinical outcome in the cirrhotic patient.
The clotting process is a dynamic array which can be explained as occurring in four phases:
- •
Platelet plug initiation and formation.
- •
Clotting process propagation by coagulation cascade.
- •
Clotting termination by antithrombotic mechanisms.
- •
Clot removal by fibrinolysis.
The liver plays a central role in each of these phases. As expected, cirrhotic patients have a disturbed balance between pro-coagulant and anti-coagulant factors, and a very poor reserve of these factors leads to increased bleeding risk as well as increased thrombotic risk. This coagulation imbalance is not reflected by conventional coagulation test.
Although variceal bleeding remains being the most prevalent coagulopathy-associated exacerbation, thrombotic complications are not uncommon in the cirrhotic patient1–4 (Figure 1, Table 1).

Coagulation abnormalities according to cirrhosis progression, reflected by Child-Pugh Score. As cirrhosis progress, levels of tissue factor pathway inhibitor, protein C, antithrombin, factors II, V, VII, IX, X, plasminogen activator inhibitor, platelet count and TAFI decreases, while tissue factor, thrombomodulin, von Willebrabd Factor, homocysteine and platelet activation increases, leading to stable thrombin generation and a fragile balance between thrombosis and bleeding. Aggregated factors, such as portal hypertension may trigger bleeding or thrombotic events. TAFI: Thrombin-activatable fibrinolysis inhibitor.
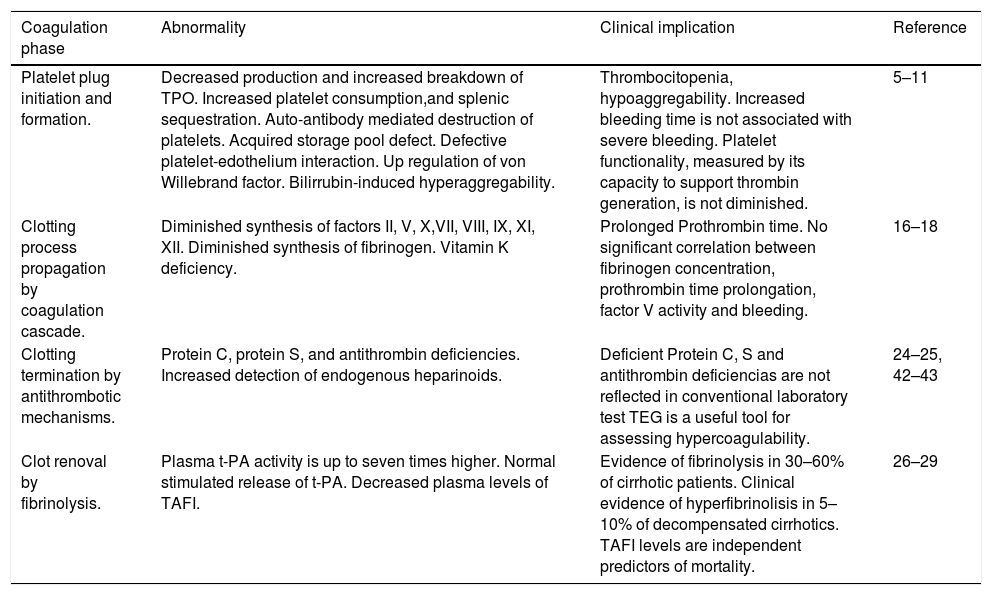
Coagulation abnormalities in cirrhotic patients.
| Coagulation phase | Abnormality | Clinical implication | Reference |
|---|---|---|---|
| Platelet plug initiation and formation. | Decreased production and increased breakdown of TPO. Increased platelet consumption,and splenic sequestration. Auto-antibody mediated destruction of platelets. Acquired storage pool defect. Defective platelet-edothelium interaction. Up regulation of von Willebrand factor. Bilirrubin-induced hyperaggregability. | Thrombocitopenia, hypoaggregability. Increased bleeding time is not associated with severe bleeding. Platelet functionality, measured by its capacity to support thrombin generation, is not diminished. | 5–11 |
| Clotting process propagation by coagulation cascade. | Diminished synthesis of factors II, V, X,VII, VIII, IX, XI, XII. Diminished synthesis of fibrinogen. Vitamin K deficiency. | Prolonged Prothrombin time. No significant correlation between fibrinogen concentration, prothrombin time prolongation, factor V activity and bleeding. | 16–18 |
| Clotting termination by antithrombotic mechanisms. | Protein C, protein S, and antithrombin deficiencies. Increased detection of endogenous heparinoids. | Deficient Protein C, S and antithrombin deficiencias are not reflected in conventional laboratory test TEG is a useful tool for assessing hypercoagulability. | 24–25, 42–43 |
| Clot renoval by fibrinolysis. | Plasma t-PA activity is up to seven times higher. Normal stimulated release of t-PA. Decreased plasma levels of TAFI. | Evidence of fibrinolysis in 30–60% of cirrhotic patients. Clinical evidence of hyperfibrinolisis in 5–10% of decompensated cirrhotics. TAFI levels are independent predictors of mortality. | 26–29 |
TPO: thrombopoietin. TEG: thromboelestography.
Cirrhotic patients commonly present quantitative and qualitative platelet defects. Thrombocytopenia may results from decreased Thrombopoietin (TPO) production and/or increased TPO breakdown, myelosuppression (resulting from acute hepatitis C infection, folic acid deficiency, or ethanol toxicity), splenic platelet sequestration, increased platelet consumption resulted from sustained low-grade disseminated intravascular coagulation or auto-antibody mediated destruction of platelets.5–8
The existence of a functional platelet defect (hypoaggregability) was described by Thomas in 1967.9 Bleeding time prolongation correlates weakly with platelet count. Qualitative platelet defects described in cirrhotic patients includes:5–10
- •
Acquired storage pool defects, such as decreased Adenosin triphosphate (ATP) and serotonine in dense granules.
- •
Decreased thromboxane A2 (TXA2) synthesis.
- •
Altered transmembrane signal transduction: quantitatively decreased glycoprotein Ib and aIIb3 receptors (as a consequence of proteolysis by the overactive fibrinolytic system), decreased response to collagen, thrombin, arachidonic acid, adenosin diphosphate (ADP), epinephrine, ristocetin.
- •
Extrinsic factors such as abnormal high-density lipoprotein and apolipoprotein E levels, reduced hematocrit and increased levels of platelet inhibitors (such as endothelium-derived nitric oxide and prostacyclin). Defective platelet-endothelium interaction may exist, and additive effect of concomitant uremia-related, sepsis related or drug related platelet dysfunction might also be present.
There are also factors that promote platelet activation e.g. von Willebrand factor (vWF) that is reported to be upregulated in cirrhotic patients. Unconjugated bilirubin is a strong inducer of platelet aggregation similar to adenosine diphosphate (ADP) in isolated platelets.11,12 Tenfold concentrated ascitic fluid causes irreversible platelet aggregation and serotonin release of normal platelet-rich plasma similar to collagen.13 In cholestatic liver disease, platelets demonstrate a hyperaggregability; thrombosis of portal veins has also been detected in 40% of primary biliary cirrhosis (PBC) liver at the time of transplantation.10 The bleeding time is prolonged in up to 40% of patients who have liver disease and it can be explained only in part by a concomitant thrombocytopenia, in fact, there is a weak correlation between bleeding time, platelet count and bleeding risk.14
Desmopressin, an analogue of vasopressin, shortens the bleeding time in these patients, by enhancing endothelial-derived vWF level, but it has shown no efficacy in controlling variceal bleeding or reducing blood loss in patients undergoing liver transplantation or liver resection. This indicates that normalization of bleeding time does not necessarily diminish bleeding risk, and bleeding time is not a reliable prognostic factor of significant bleeding. The platelet function analyser-100 (PFA-100) is a rapid new in vitro test that provides a quantitative measure of primary hemostasis at high shear stress using citrated whole blood. This automated device functions with blood flowing under a constant vacuum through a capillary and a microscopic aperture in a membrane coated with collagen and agonists. As a result, the closure time of the aperture is a measurement of platelet adhesion/aggregation. Prolonged closure time can be corrected by elevating the hematocrit in the blood of patients with liver disease, suggesting that the influence of hematocrit (and other physiologic factors) on platelet function is probably more relevant than the intrinsic platelet defects commonly found in patients who have liver disease.8
The clinical relevance of thrombocytopenia and hypoagregability in cirrhosis remains unclear: Thrombocytopenia is mostly mild to moderate, and it is not associated with severe bleeding in patients with stable liver disease, and the clinical relevance of thrombocytopenia in most patients who have liver disease has a questionable significance. Platelet functionality, as measured by its capacity to support thrombin generation, is not diminished in patients who have stable cirrhosis: under physiologic conditions of flow, platelets from patients who have liver cirrhosis are able to interact normally with collagen and fibrinogen as long as the platelet count and hematocrit are adjusted to the levels found in healthy subjects.10,14,15
Clotting Process Propagation by the Coagulation CascadeThe majority of procoagulant factors, of the coagulation cascade, such as fibrinogen and factors II, V, X, VII, VIII, IX, XI, XII are synthesized in the liver, therefore, when synthetic liver function fails, procoagulant factors fail in parallel to the degree of liver failure. FV and FVII have the shortest in vivo half-lives (12 and 4–6 h, respectively) and are the most decreased procoagulants in patients with liver disease.16
Patients with liver disease may have vitamin K deficiency due to poor nutrition, or malabsorption caused by decreased bile production or biliary obstruction. The prevalence of vitamin K deficiency (defined as low plasma vitamin-K1 levels measured by reverse-phase high performance liquid chromatography) in patients with histologically proven PBC is 23.3%. These results do not correlate with the vitamin- K deficiency prevalence when prothrombin time (PT) prolongation is used as a surrogate marker, and therefore PT should not be assumed to be a reliable marker of vitamin K deficiency in patients with PBC.17,18
FVIII (synthesized in the liver and endothelial cells) and vWF (present in endothelial cells, platelets and megakaryocytes),19–22 are also “acute phase reactants” thus normal or elevated factor VII and vWF levels may be found in cirrhotic patients caused by continued endothelial cell FVIII production despite decreased hepatic FVIII production, and increased synthesis caused by acute disease or inflammation. Changes in the latter result from compensatory increased production, decreased levels of a vWF cleaving protein in cirrhotics, and an increased presence of vWF on endothelial walls due to a chronic inflammatory state.16,19–23
Because PT is dependent on FVII levels and FVII has the shortest in vivo half-life, an isolated prolonged PT is common for patients with liver disease. Although it is important to know the expected procoagulant synthetic and coagulation test profile in cirrhotic patients, the most important question is whether or not clinical bleeding can be predicted based on the concentration of procoagulant factors (i.e. prolongation of procoagulant laboratory tests) the prolongation of procoagulant laboratory tests. Some insight into this problem is provided by the study from hemophilias A and B, in which patients with more than 6% of the normal activity level of a given coagulation factor is enough to prevent spontaneous bleeding, but this single factor coagulation deficiency model may not be applicable to cirrhosis, in which almost every coagulation factor is deficient. Until now, no significant correlation has been found between fibrinogen concentration, PT prolongation, factor V activity and bleeding tendency clinical markers such as melena, hematemesis, variceal bleeding, gastric ulcer bleeding, persistent skin bleeding after liver biopsy, menorrhagia, positive fecal occult blood test and blood transfusion requirements.16
Clotting Termination by Antithrombotic MechanismsPatients with liver disease commonly have decreased levels of proteins C, S, and antithrombin. Proteins C and S are vitamin-K-dependent proteins synthesized by hepatocytes. Protein C, protein S, and antithrombin deficiencies observed in patients with liver disease have been attributed to decreased hepatocyte function, increased consumption due to disseminated Intravascular coagulation (DIC) or hyperfibrinolysis, and vitamin K deficiency.16
Infection has been associated with increased detection of endogenous heparinoids, probably as a reflection of endothelial dysfunction and associated changes in the endothelial glycocalyx, possibly mediated by infection-related changes in nitric oxide metabolism. Glycosaminoglycans (heparinoids) found in the vessel wall are bound by the endothelium and help to maintain hemostatic balance by preventing clot formation and facilitating blood flow within a vessel. Two specific glycosaminoglycans (GAGs), heparan sulphate and dermatan sulphate, have anticoagulant properties similar to heparin. Increased levels of these proteins, measured by thromboelastography (TEG), have been found in patients with cirrhosis, and even higher concentrations were found in subjects with cirrhosis and active bleeding or infection.24,25
Clot Removal by FibrinolysisClot and fibrin degradation is determined by the endothelial release of tissue plasminogen activator (t-PA) and its subsequent inhibition by plasminogen activator inhibitor type-1 (PAI-1). To maintain a normal fibrinolytic balance, increases in plasma t-PA concentrations are associated with compensatory increases in plasma PAI-1 concentrations. PAI-1 forms a complex with t-PA to cause an overall reduction in free tPA “activity”. In patients with cirrhosis, plasma t-PA activity is up to seven times higher, and it is proportional to the severity of cirrhosis and may contribute to the risk of variceal hemorrhage by the removal of fibrin clot at the site of vascular injury. Patients with alcoholic cirrhosis have a higher basal plasma t-PA antigen concentration but a normal stimulated release of t-PA. However, because of a failure to increase plasma concentrations of its inhibitor PAI–1, plasma t-PA activity is elevated during both basal and stimulated t-PA release.26,27
According to the degree of liver dysfunction, evidence of fibrinolysis may be found in 30 to 60% of patients with chronic liver disease, and clinically evident hyperfibrinolysis has been estimated to occur in 5 to 10% of those with decompensated cirrhosis. Hyperfibrinolysis promotes clot dissolution and decrease platelet aggregation due to degradation of vWF and fibrinogen platelet receptors.28,29
TAFI (thrombin activatable fibrinolysis inhibitor) is a plasmatic proenzyme synthesized in the liver that, upon activation by thrombin or other enzymes (plasmin, trypsin, neutrophil elastase), inhibits fibrinolysis by removing carboxy-terminal lysine residues from partially degraded fibrin. Thereby reducing plasmin formation TAFI is an important regulator of physiological fibrinolysis and alterations in TAFI-activating mechanisms or in TAFI levels contribute to bleeding or thrombotic manifestations.27
Decreased plasma levels of TAFI have been reported in patients with acute or chronic hepatocellular diseases of different aetiology, and the degree of TAFI levels is proportional to the severity of the disease. TAFI deficiency is probably, related with increased fibrynolysis and impaired thrombin generation. In a prospective follow up study including 65 patients with liver cirrhosis with a mean follow up of 816 days, lower TAFI levels, were strongly correlated with fibrinolysis time, and TAFI levels were lower in non-survivors, but fibrinolysis time was not significantly related to survival. TAFI levels were an independent predictor of mortality after correction for age, sex, aetiology of cirrhosis, and Child-Pugh class.27
Bleeding and Thrombotic Events in the Cirrhotic PatientCirrhosis accounts an increased risk for both bleeding and thrombotic episodes. Bleeding episodes in cirrhotic patients may be classified as spontaneous (variceal, non-variceal gastrointestinal bleeding and cerebral hemorrhage) or procedure-related (including biopsy, central vein cannulation and paracentesis). Thrombotic episodes are categorized according to the affected anatomic region (e.g. deep vein thrombosis, pulmonary embolism).30–31
- •
Spontaneous bleeding episodes:
- a)
Variceal bleeding. Variceal bleeding occurs in 20–30% of cirrhosis patients and accounts for 80–90% of bleeding episodes in these patients, 30% of initial bleeding episodes are fatal and as many as 70% of survivors have recurrent bleeding after two weeks from the first episode. Gastrointestinal bleeding is mainly haemodynamic in its mechanisms and is related to portal hypertension. Risk factors for developing variceal bleeding include advanced Child-Pugh class, large varices and the presence of red wale markings. Constipation, vomiting, severe coughing, excessive consumption of alcohol and bacterial infections may also contribute to variceal bleeding. High levels of D-dimer and t- PA have been described as significant laboratory markers of risk of variceal bleeding, independently of other laboratory markers and severity of liver disease. In decompensated cirrhotic patients, variceal bleeding is also the main coagulopathy-related problem, representing 50% of all hemorrhagic events.4,31
- b)
Intracerebral haemorrhage. The prevalence of spontaneous intracerebral hemorrhage in hospitalized cirrhotic patients, is low (0.8% for all aetiologies, 80.3% in virus-related and 1.8% in alcohol-related groups), and it is reported not to be related to Child-Pugh score or prolonged PT.31 While a retrospective population-based case-control study found an increased risk of intracerebral hemorrhage for both alcoholic liver and non-alcoholic liver cirrhosis patients (adjusted OR of 4.8 and 7.7 respectively), a five year cohort study found no increased risk of hemorrhagic stroke in patients with non alcohol-related cirrhosis after adjusting for the patients’ geographical location, hypertension, diabetes, coronary heart disease, heart failure, atrial fibrillation and hyperlipidaemia.32,33 Alcohol-related cirrhotic patients have the shortest time interval between diagnosis of liver disease and hospitalization due to brain hemorrhage (2.3 years vs. 6.2 years in non-alcoholic cirrhosis) and heavy drinkers had a significantly higher risk of hemorrhagic stroke than non-heavy drinkers. In some cases, cerebral metastasis bleeding may be the initial manifestation of hepatocellular carcinoma in cirrhotic patients.31
- a)
- •
Procedure-related bleeding episodes:
- a)
Biopsy-associated bleeding. The incidence of biopsy-associated bleeding in patients with histological diagnosis of cirrhosis is 0.7%; neither PT nor prolonged Activated partial thromboplastin time (aPTT) and thrombocytopenia are reliable predictors of biopsy related bleeding.31 In a multicenter study of liver biopsy-related complications in patients with advanced hepatitis C that involved 2,740 percutaneous biopsies performed over 7 years (80% performed with ultrasound guidance, 40% performed by aspiration needle, and 60% used a cutting needle) the bleeding risk was 0.4% for patients with an international normalized ratio (INR) of 1.1 or less, 1.1% for patients with an INR of 1.2, and 2.4% for patients with an INR of 1.3 or greater; however, 37.5% of the patients who had bleeding had INR < 1 and none of the patients under-going a biopsy with INR values greater than 1.5 experienced bleeding. Other factors that were statistically different between those with bleeding and those without were lower albumin, presence of varices, platelets less than 60,000.34
- b)
Central venous canulation. Bleeding complications during central venous canulation in cirrhotic patients are rare and can be safely performed in patients with liver disease and abnormal coagulation tests, even if the INR is 1.5 or more. Although the occurrence of severe bleeding complications is very low for both for subclavian and internal jugular routes of access, in order to prevent minor bleedings and vascular complications, cannulations should to be performed by experienced practitioners with ultrasound guidance.31
- c)
Paracentesis. Paracentesis has a 0.2% incidence of severe haemorrhage, with a death rate of 0.016%. Paracentesis-related bleeding risk has not been found to be associated with a PT prolonged up to twice the midpoint of normal or with a platelet count of < 50,000.31
- a)
As mentioned above, not only bleeding but also thrombosis complicates the clinical course of cirrhosis, mostly venous thromboembolism (VT), and portal vein thrombosis (PVT).
- •
Venous thromboembolism. Patients with cirrhosis have been found to have a 1.65–1.74 relative risk of VT when compared with controls. The prevalence of VT in hospitalized cirrhotic patients is 0.8%, and 0.5% of hospitalized cirrhotic patients present their first venous thrombosis during hospitalization. Reduced aPTT, and low serum albumin levels have been found to be an independent predictor of developing VT, while prolonged INR did not appear to protect against the development of hospital-acquired VT.30,35 Shah, in a prospective study on decompensated cirrhotic patients admitted to a inpatient Hepatology Service described that, over a 6 month period, 7% of the patients presented DVT.4
- •
Portal vein thrombosis and pulmonary embolism. PVT is the most common thrombotic event in cirrhotic patients, with a reported incidence of 7.4 to 11%. Reported risk factors include prothrombotic states arising from protein C, protein S and antithrombin III deficiencies, myeloproliferative disorders, sclerotherapy of esophageal varices, abdominal surgery, hepatocellular carcinoma, antiphospholipid syndrome, mutations of prothrombin (PTHR G20210A), factor V Leiden (FVL G1691A) and methylenetetrahydrofolate reductasa, a key enzyme in the folate cycle and homocysteine metabolism (MTHFR C677T).35,36
Northup, in a case control study that included 113 cirrhotic patients reported a 0.5% incidence of deep vein thrombosis (DVT) /Pulmonary embolism (PE). From all thrombotic events registered, 65.5% corresponded to DVT 19.5% corresponded to PE, 15% to patients with both DVT and PE, and only 7% of the patients who presented thrombosis were receiving medical thromboprophylaxis at the time the thrombotic event occur.37 Garcia-Fuster, in a retrospective study that included 2074 cirrhotic patients the DVT/PE incidence were 0.8%; 59% of the events corresponded to DVT, 35% toPE and 6% corresponded to patients with both DVT and PE.38
Special Laboratory Evaluation of Coagulation in Cirrhotic PatientsThe PT and aPTT are used ostensibly to investigate patients who have cirrhosis even though these tests are known to be poor predictors of bleeding in this category of patients because cirrhosis is a condition in which levels of such naturally occurring anticoagulants as protein C and antithrombin are reduced in parallel with the procoagulants, and PT and aPTT are responsive only to the thrombin generated as a function of procoagulants but are much less responsive to the inhibition of thrombin mediated by the anticoagulants. The PT also has been used over the years in combination with other clinical and laboratory parameters to calculate such prognostic indexes as the Child-Pugh or the model of end-stage liver disease (MELD).39,40
TEG allows continuous registration of the blood viscoelastic changes upon activation by cephaline or tissue-factor plus calcium-chloride: the variables analyzed by this test includes:41
- •
The R value (reaction time, clotting time), represents the time until the first evidence of a clot is detected.
- •
The K value (clot formation time), is the time from the end of R until the clot reaches 20 mm, representing the speed of clot formation.
- •
The angle is the tangent of the curve made as the K is reached and offers similar information to K value.
- •
The maximum amplitude, maximum clot firmness is a reflection of clot strength.
A mathematical formula determined by the manufacturer can be used to determine a Coagulation Index, an overall assessment of coagulability.41
In a study where citrated blood samples from 51 adult cirrhotic patients were submitted to thromboelastographic study, 27% had abnormal clot time, 80% presented abnormal clot formation time and 76% had abnormal maximum clot firmness. Clot formation time and maximal clot firmness were correlated with the platelet-count, antithrombin and fibrinogen, PT was correlated with K value and maximal amplitude, and none of the coagulation parameters were correlated with R value. The correlation of the Child-Pugh-score versus maximal clot firmness or PT was −0.457 (p < 0.001) or 0.484 (p < 0.001), suggesting that maximal clot firmness may be a suitable prognostic index.42
TEG has also been used to provide evidence for the generation of endogenous heparinoids as possible contributors to the coagulopathy in patients who have liver disease. In this application, TEG using native blood could be useful in clinical practice to detect the anticoagulant effect of endogenous heparinoids and the associated hemorrhagic events.41 In a study of the prognostic value of TEG as a rebleeding predictor in cirrhotic patients with variceal hemorrhage, those who present rebleeding were more hypocoagulable before the day of rebleeding as shown by longer R (42 v 24 mm, p < 0.001) and K (48 v 13 mm, p < 0.001) and smaller angle (12 v 38 degrees, p < 0.001) compared with the mean of daily results of the non-rebleeding group. Routine coagulation test, however, showed no significant differences between the two groups.43
Therapeutic Approaches to Thrombotic ComplicationsSince the recognition that cirrhosis does not represent a self-autoanticoagulation state (but rather a mixure of both prothrombotic and antithrombotic anomalies), it has been evident that, even in the presence of abnormal coagulation tests, cirrhotic patients may need thromboprophylaxis or anticoagulation in certain clinical scenarios, especially those involving an additional thrombotic risk (e.g. venous stasis, infection, congestive heart failure, acute respiratory disease, and surgery).44
Bechmann evaluated the pharmacokinetics of LMWH in 84 consecutive patients with cirrhosis and a clinical indication for prophylactic or therapeutic anticoagulation. Antifactor Xa activity was negatively correlated with the severity of the liver disease, and a positive correlation was observed between antithrombin-III levels and antifactor-Xa value. Antithrombin-III itself was negatively correlated with the severity of liver disease. Seven patients had an episode of variceal bleeding. No patient died during the observation interval and no thromboembolic events occurred. These results suggest that prophylactic use of low molecular weight heparin (LMWH) in cirrhotic patients is safe. A decreased anti-Xa value in cirrhotic patients and a negative correlation with liver function challenge the unconditional use of anti-Xa assays in LMWH monitoring in cirrhotic patients and reveals a potential limitation of anti-Xa analysis in these patients. Low levels of antithrombin, because of reduced hepatic synthesis, are the most likely cause of this phenomenon. Cirrhotic patients have also been found to have an increased response to LMWH, which correlates with the severity of liver disease, despite reduced antithrombin and anti-Xa activity levels.45,46
The factor Xa inhibitors rivaroxaban and apixaban are metabolized in the liver and they are contraindicated in severe hepatic diseases because their metabolic inactivation is impaired. Metabolic conversion of the prodrug dabigatran etexilate to dabigatran, a thrombin inhibitor, is completed in the liver and followed by partial biliary excretion of a conjugated derivate. Idraparinux, an antithrombin dependent FXa inhibitor, has no hepatic clearance, but its long half-life (approximately 80 h) and the lack of antidote do represent major problems if bleeding occurs. These drugs must be use with caution or are contraindicated in the presence of renal failure.47
In respect to the safety and efficacy of anticoagulation for the treatment of thrombotic complications of cirrhosis, Senzolo48 found that, among 33 patients with PVT treated with LMWH (95 anti-Xa U/Kg body weight tid), 12 (36.36%) reached complete repermeation and 9 (27.27%) reached partial repermeation only, and only 2 patients (6%) experienced complications (1 patient developed heparin-induced thrombocytopenia and another experienced non variceal bleeding).
In a study of 55 cirrhotic patients with PVT, Delgado49 described the clinical outcomes after anticoagulation treatment in 47 patients with PVT and had received anticoagulation treatment. LMWH was the initial treatment in 47 patients: of them, 21 were shifted to vitamin K antagonist (VKA) after 17 days; 8 patients received VKA to reach a target INR of 2.5 as the initial treatment: 25 patients reached total repermeation, 8 reached partial repermeation, 5 patients presented non variceal bleeding and 6 presented variceal bleeding.
In cirrhotic patients with acute variceal bleeding and PVT, anticoagulation with LMWH administered after hemostasis is achieved (by band ligation, repeated band ligation or injection sclerotherapy combined with argon plasma coagulation) has reported to achieve complete recanalization of the portal vein within 2–11 days, with no significant increase in rebleeding incidence. Even after resection of hepatocarcinoma, thromboembolism has been reported to be higher between cirrhotic patients who do not receive thromboprophylaxis, and thromboprofilaxis with LMWH has not shown any increase the rela tive risk of postoperative bleeding.50,51
Villa52 in a randomized non-blinded single-center controlled trial, evaluated the safety and efficacy of enoxaparin versus no treatment, in preventing PVT in patients with advanced cirrhosis. Patency of portal vein was evaluated by ultrasound every three months and by computed tomography every six months. After 48 weeks of follow up, none of the patients receiving enoxaparin had developed PVT, compared with 6 of 36 (16.6%) of controls (P = 0.025). At 96 weeks, no patient developed PVT in the enoxaparin group, compared with 10 of 36 (27.s7%) controls (P < 0.001). The incidence of new onset descompensation was not statistically different between the enoxaparin and control group. Enoxaparin treated group. showed a trend toward amelioriation in renal function biomarkers, liver function test and higher survival rates. The incidence of thrombocytopenia and esophageal variceal bleeding shown no significant difference between enoxaparin-treated and control groups.
Amitrano53 evaluated the safety and efficacy of LMWH to treat non-neoplastic PVT in cirrhotic patients who were suitable for liver transplantation, and did not had any active source of bleeding (endoscopic examination of varices and betablockade was started prior to the administration of LMWH). Once PVT was documented with ultrasound and computed tomography, LMWH al 200 U/kg/day was started and continued for at least six months. Patients were submitted to Doppler ultrasound examination monthly. Ultrasonographic follow-up was performed monthly and computed tomography was performed at sixth month. At six months complete recanalization occurred in 33.3%, partial recanalization in 50% and 16.7% of patient had no response.
Among patients who had achived partial recanalization, 65% reached complete recanalization after continuing anticoagulation for 7 to 17 months. Overall, complete recanalization was achived in 75% of patients in a median time of 11 months. No patient presented severe side effects which requires interruption of therapy. Until now, the results obtained from published series on anticoagulation therapy for PVT thrombosis in cirrhotic patients have shown that anticoagulation is effective in achieving portal vein repermeation and does not increase the risk of mayor complications (Table 2).
Published series on anticoagulation therapy for portal vein thrombosis prevention and treatment in cirrhotic patients.
| Reference | Study design | Anticoagulation strategy | Portal patency achieved (%) | Anticoagu lation-related complications (%) |
|---|---|---|---|---|
| Delgado49 | Retrospective analysis. 55 patients who received anticoagulation for PVT. | Initial therapy LMWH: 47 patients. 21 shifted to VKA after 17 days. Initial therapy with VKA: 8 patients. | Complete repermeation: 25 patients (45.45%). Partial repermeation: 8 patients (14.54%). No repermeation: 22 patients (40%). | Non-variceal bleeding: 5 patients (9.09%). Variceal bleeding: 6 patients (10.90%). |
| Maruyama50 | Prospective study. LMWH for PVT in patients with for variceal bleeding. 23 patients with variceal bleeding with or without PVT. Five patients had PVT and achived hemostasis. | LMWH 75 IU/kg/day after achieving hemostasis. | Complete repermeation: 5 patients (100%). | None. |
| Villa52 | Randomized controlled trial. LMWH for prevention of PVT. 34 Enoxaparin-treated patients. 36 control patients. | Enoxaparin 4,000 IU/day for 48 weeks. | At 48 and 96 weeks, 0 LMWH-treated patient (0%) developed PVT. At 48 weeks, 6 patients (16.6%) of the control group had developed PVT (p = 0.025). At 96 weeks, 10 patients (27.7%) of the control group had developed PVT (p = 0.001). | None. |
| Amitrano53 | Prospective evaluation. LMWH fornon-neoplastic PVT. 28 patients with non-neoplastic PVT. | Enoxaparin 200 U/kg/day. | Complete repermeation 21 patients (75%). Partial repermeation: 2 patients (7.14%). No repermeation: 5 patients (17.85%). | None. |
| Francoz62 | Prospective evaluation. LMWH in patients with PVT awaiting for transplantantion. 19 nandroparin-acenocumarol treated patients. 10 historical control patients. | Nadroparin, 5,700 UI/day, followed by acenocoumarol, target INR range 2–3. | Complete repermeation: 8 patients (42.1%) No change in PVT: 10 patients (52.63%). Expansion of PVT despite anticoagulation: 1 patient (5.26%). Repermeation in control group: 0 patients (0%) (p = 0.002). | None. |
| Senzolo63 | Prospective evaluation, LMWH on patients with non malignant PVT. 33 nandroparin treated patients. 21 control patients. | Nandroparin, 95 anti-Xa U/Kg tid. | Complete repermeation 12 patients (36.36%). Partial repermeation: 9 patients (27.27%). Thrombus progression: 3 patients (9.09%) Thrombus progression: 15 (71.42%) non-anticoagulated patients (p < 0.001). | Non variceal bleeding: 1 patient (0.33%). HIT: 1 patient (0.33%). |
PVT: portal vein thrombosis.
LMWH: low molecular weight heparin.
VKA: vitamin K antagonist.
INR: international normalized ratio.
HIT: heparin-induced thrombocytopenia.
The use of plasma-based blood products has been a dogmatic treatment for the coagulopathy of liver disease. It may also represent a public health issue since blood product administered to decompensated cirrhotic patients accounts for 7.7% of the total transfused red blood cell packages, 32.4% of the fresh frozen plasma units and 13.2% of platelet units consumed in a tertiary care hospital.4
Randomized clinical trials provide little support, however, using plasma before invasive procedures, except for bleeding diathesis is clearly indicated. Although plasma is commonly used in patients who have cirrhosis with prolonged INR, injudicious use should be regarded as a risk and liability in light of the limitations of the INR and the risks associated with plasma use. Moreover, the amount of fresh frozen plasma requested is typically inadequate to reach the desired INR in the great majority of cirrhotic patients. Morover, transfusion-related complications rate has been reported to be 15% at 48 h after transfusion. Described transfusion-related complication in cirrhotic patients include transfusion related acute lung injury and acute kidnay injury related to tubular necrosis.4,54,55
Adequate thrombin production seems to be present in cirrhosis when platelet counts are approximately 50,000 to 60,000/cc3, whereas optimal levels are seen with levels of 100,000/cc3 or more. Thus recommended platelet levels of at least 50,000/cc3 for moderate-risk procedures (ie, liver biopsy) and closer to 100,000/cc3 in very–high-risk procedures (ie, intracranial pressure monitor placement).15,54–55
Rheologic studies have indicated that normal platelet flow (at the periphery of the circulating blood stream where they are closer to potential binding sites in the event of vascular breach) is adversely affected with hematocrit levels less than 25%, sup porting the recommendation to maintain this level in cirrhotic patients experiencing bleeding or about to undergo a high-risk procedure. While portal hypertension plays a central role on the bleeding risk, infused volume should be considered when using blood products. The risk of transfusion related to acute lung injury, transfusion-related volume overload and other transfusion related adverse events, should be taken into account before using blood products in cirrhotic patients.54
Initial studies of desmopressin in compensated cirrhotics showed that it shortened the bleeding time and partial thromboplastin time with increases in factor VIII and vWF, but it has shown no efficacy in controlling variceal bleeding or reducing blood loss in patients undergoing liver transplantation or liver resection, indicating that normalization of bleeding time not necessarily diminishes bleeding risk, and bleeding time does is not a reliable prognostic factor of significant bleeding. The combination of desmopressin and terlipressin increased the rebleeding rate compared with terlipressin alone.56,57
Stanca,58 in a prospective, randomized controlled trial, evaluated the efficacy of intranasal desmopressin as an alternative to transfusion to correct the coagulopathy of cirrhotic patients undergoing dental extraction. The etiology of liver cirrhosis in the studied population was hepatitis C in 22 patients (61%), multifactorial in 6 (16%) alcohol in 3 (8%), and criptogenic in 2 (6.1%), hemochromatosis, primary sclerosing cholangitis and hepatitis B represented 3.1% each. No patient receiving desmopressin had significant bleeding post procedure; in comparison, 1 patient in the transfusion group bled and required further transfusion, and another patient experienced an allergic reaction. In addition, treatment associated average costs were lower for desmopressin ($700/patient) compared with transfusion ($1,173/patient) group.
Several formal studies have evaluated the role of factor VII in the control of bleeding during variceal hemorrhage, liver transplantation, and partial hepatectomy. None of these reports demonstrated efficacy of factor VII on the composite endpoint of failure to control bleeding within 24 h, failure to prevent clinically significant rebleeding or death within 5 days of first dosing. However, subanalyses demonstrated significant changes in the composite endpoint within upper gastrointestinal hemorrhage in patients with Child-Turcotte-Pugh class B and C cirrhosis, higher survival, and reduced transfusion requirements at the time of transplant. Further studies are warranted with Factor VII in light of recent advances in understanding of coagulation disorders in order to assess its efficacy in more specific situations (i.e. avoid its use in conditions of hypercoagulability).59–62
ConclusionsThe liver is a key element of the coagulation system. Patients with cirrhotic liver disease fail to synthesize both prothrombotic and antithrombotic factors. In addition, endothelial damage, endothelial dysfunction, portal hypertension, infections and renal failure may disrupt the coagulation system hemostasis in cirrhotic patients. The clinical presentation, whenever bleeding or thrombosis, depends upon the stage of cirrhosis, added triggering factors and individual factors such as pre existing prothrombotic or antithrombotic conditions. Regular coagulation test, such as INR, PT or aPTT are poor predictors of bleeding or thrombotic complications, since they not evaluate the entire coagulation system. TEG may be a useful tool for evaluating coagulation abnormalities in cirrhotic patients, since it takes into account primary hemostasis, coagulation, and fibrinolysis. Therapeutic approach should include both thrombotic and bleeding events, and should not be directed in achieving normal laboratory coagulation test results.
Abbreviations- •
ADP: adenosin diphosphate.
- •
aPTT: activated partial thromboplastin time.
- •
ATP: adenosin triphosphate.
- •
DIC: disseminated intravascular coagulation.
- •
DVT: deep vein thrombosis.
- •
GAGs: glycosaminoglycans.
- •
HIT: heparin-Induced thrombocytopenia.
- •
ICU: Intensive Care Unit.
- •
INR: international normalized ratio.
- •
LWMH: low molecular weight heparin.
- •
MELD: model of end-stage liver disease.
- •
PAI-1: Plasminogen activator inhibitor type-1.
- •
PBC: primary biliary cirrhosis.
- •
PE: pulmonary embolism.
- •
PFA-100: the platelet function analyser-100.
- •
PT: prothrombin time.
- •
PVT: portal vein thrombosis.
- •
TAFI: thrombin-activatable fibrinolysis inhibitor.
- •
TEG: thromboelestography.
- •
t-PA: tissue plasminogen activator.
- •
TPO: thrombopoietin.
- •
TXA2: thromboxane A2.
- •
VKA: vitamin K antagonist.
- •
VT: venous thromboembolism.
- •
vWF: von Willebrand factor.



