





Bile acids (BAs) regulate the absorption of fat-soluble vitamins, cholesterol and lipids but have also a key role as signaling molecules and in the modulation of epithelial cell proliferation, gene expression and metabolism. These homeostatic pathways, when disrupted, are able to promote local inflammation, systemic metabolic disorders and, ultimately, cancer. The effect of hydrophobic BAs, in particular, can be linked with cancer in several digestive (mainly oesophagus, stomach, liver, pancreas, biliary tract, colon) and extra-digestive organs (i.e. prostate, breast) through a complex series of mechanisms including direct oxidative stress with DNA damage, apoptosis, epigenetic factors regulating gene expression, reduced/increased expression of nuclear receptors (mainly farnesoid X receptor, FXR) and altered composition of gut microbiota, also acting as a common interface between environmental factors (including diet, lifestyle, exposure to toxics) and the molecular events promoting cancerogenesis. Primary prevention strategies (i.e. changes in dietary habits and lifestyle, reduced exposure to environmental toxics) mainly able to modulate gut microbiota and the epigenome, and the therapeutic use of hydrophilic BAs to counterbalance the negative effects of the more hydrophobic BAs might be, in the near future, part of useful tools for cancer prevention and management.
Lipids in bile include three species: bile acids (BAs), cholesterol and phospholipids. “Primary” BAs are synthe-tized in the liver from cholesterol as cholic acid (CA) and chenodeoxycholic acid (CDCA) starting from the host cytocrome P450 family enzymes (more than 200 enzymes) via the “classical” (CYP7A1) and “alternative” (CYP27A1) BA synthetic pathways (involving at least 14 enzymes). BAs in the liver are then conjugated to taurine and glycine (bile acid cholyl-CoA synthetase [BAC] activity and ami-dation at C24 to either glycine or taurine by the enzyme bile acid-CoA:amino acid N-acyltransferase [BAT]),1 to be secreted as more hydrophilic molecules into the bile and stored and concentrated in the gallbladder, where the aqueous bile undergoes water reabsorption and concentration.2,3 The gallbladder is stimulated mainly after a meal by the entero-hormone cholecystokinin (CCK) and this step leads to biliary secretion of concentrated, BA-enriched bile into the duodenum which will flow down to the ile-um and the colon. Most of the BAs will be actively reab-sorbed as conjugated BAs in the terminal ileum and return to the liver through the portal blood circulation. About 15% of conjugated BAs will escape the terminal ileum absorption and will enter the colon; the resident microbiota will provide deconjugation of the BA steroid nucleus from the amide-bond taurine and glycine (by the bile salt hy-drolases, BSH) and further biotransformation of unconju-gated primary hepatic BAs (CA, CDCA) into secondary (deoxycholic acid, DCA and lithocholic acid, LCA) and tertiary bile acids (ursodeoxycholic acid, UDCA).4 The bile salt hydrolase (BSH) enzymes are essential in this respect. Both conjugated and unconjugated BAs will reach the liver and after liver uptake, the secondary BAs are reconjugated with taurine and glycine to complete the BA pool (i.e. glyco, tauro- CA, CDCA, DCA, LCA, UDCA).5 Only ≈ 5% (i.e. 0.2-0.6 g per day) of the BA secreted are lost in feces, and this portion equals to the amount of hepatic synthesis (0.2-0.6 g/day). The overall pool is therefore calculated from 3 g of BA undergoing 4-12 cycles per day = 12-36 g/day.5
BAs are able to regulate their own synthesis via at least two negative feedback mechanisms:
- 1)
In the hepatocyte, binding of BAs to FXR in the nucleus will activate the formation of the FXR/RXR het-erodimer and synthesis of the inhibitory SHP, which will inhibit the activity of the liver receptor homolo-gous-1 (LRH-1) and CYP7A1 transcription.6,7
- 2)
In the enterocytes, the activation of FXR leads to the secretion of the enterokyne FGF15/19, activation of FGFR4 tyrosine kinase/β-klotho (a coexpressed membrane-bound glycosidase) signaling in the hepatocyte basolateral plasma membrane.8,9 The JNK-mediated pathway will suppress CYP7A1 transcription.5,10,11
BAs play complex key roles in health and disease, as recently pointed out by Volle D.H.12
The physiologic functions of BAs include the intestinal solubilization and absorption of fat-soluble vitamins, cholesterol and lipids.13 However, the role of BAs in human metabolism goes beyond that of pure fat emulsifier, because of their chemical moieties as soluble amphiphiles. BAs also have distinct roles as signaling molecules with metabolic effects via interaction with the nuclear receptors farnesoid X receptor (FXR), pregnane X receptor (PXR), and vitamin D receptor (VDR), G-protein coupled receptors such as the G-protein-coupled bile acid receptor-1 (GPBAR-1, also known as TGR5), and cell signaling pathways such as JNK and ERK.14 Through these interactions, BAs help regulate nutrient metabolism of energy, glucose, lipid and lipoprotein.13,15-17
The overall hydrophobicity scale of BA, which is directly related to cytotoxicity is the following: UDCA < CA < CDCA < DCA < LCA. However, for BA-FXR interaction the rank order of potency is estimated to be CDCA > LCA = DCA > CA both in the conjugated and unconjugated forms18 and for BA-GPBAR-1 interaction the rank order of potency is estimated to TLCA > TDCA > TCDCA > TCA.13 Thus, subtle quantitative or qualitative perturbations of the BA pool may greatly affect several BA physiological functions in the body.1
Abnormalities in BA synthesis, secretion, absorption and local and systemic effects have been implicated during inflammation,16,19 metabolic disorders,16 liver diseases,19,20 and many other conditions.21,23 BAs play also a crucial role as potential cancer-promoting agents24-27 and in regulating the proliferation of cancer cells of diverse origin.28-31 A causal relationship between BAs (in particular DCA, one of the components of the human BA pool) and cancer was firstly proposed in 1940.32 Only in the last decades the toxic and cancerogenic effects of BAs (mainly in terms of secondary BAs) have been better elucidated.
In the current review we examine the main mechanisms linking BAs to both environmental stimuli and cancer onset/progression, in order to dissect future lines of research in primary prevention and therapy in oncology.
Bas and Cancer: General ConsiderationsPathways potentially linking BAs to cancer are being identified and involve oxidative stress with DNA damage and ge-nomic instability,33 apoptosis,34 epigenetic factors,18,35-38 activation of nuclear receptors and metabolic and cellular homeostasis,28,29,31,39-43 interactions with- and changes of gut microbiota.1,44 These mechanisms can also be secondary to environmental stimuli (i.e. diet, lifestyles, exposure to environmental toxics) and their relationships with cancer have been recognized as critical at different levels of the gastrointestinal tract (oesophagus,36,40,45 stomach,46,47 liver,48-50 pancre-as,41,42,51 biliary tract,52 colon39) and in extra-digestive organs (i.e. prostate,31,53,54 breast43,55-58). Cooperative effects with other cancer-promoting agents (i.e. alcohol,59-63 smoking,64,65 environmental pollutants66-69) are also possible. Nevertheless, recent observations suggest that some BAs might have beneficial effects as anti-cancer agents as well, while modulating the same pathways which induce toxicity, i.e. apopto-sis,70,71 clonogenic potential,54,72,73 oxidative processes underlying DNA damages.74
Direct Effects of Bas: From Oxidative Stress to Inflammation and Mutagenic ProcessesBAs have both hydrophilic and hydrophobic surfaces, are highly soluble, detergent-like amphiphilic molecules. While hydrophilic, less cytotoxic BAs play a protective role71 on gastrointestinal75-79 and liver80,81 cells, hydropho-bic BAs can be cytotoxic and can generate oxidative stress and DNA damage (genomic instability), which is a predisposing factor for cancer.24 The main general mechanisms involved are the increased intracellular production of reactive oxygen and nitrogen species,24,27,82 and the altered expression of tumour suppressor/promoting genes.47,83,84
CDCA (chenodeoxycholic acid) and DCA are able to solubilise the cell membrane and to promote immunosuppression and tissue damage.85 Dietary habits may have a role at different levels: the damaging effects of oral DCA on jejunum and colon (tissue-disrupting effect and increased permeability) are seen at concentrations induced by a high-fat diet but not by a low-fat diet and are ameliorated by administration of UDCA.76 In the liver, feeding various concentrations of BAs with diet to mice produced the following hepatotoxicity: UDCA < CA < CDCA < DCA < LCA.86 Additional mechanisms of hydrophobic BAs include the induction of apoptosis (in the short term) or apoptosis resistance (in long term)33,87 and, ultimately, the direct activation of mutagenic processes involved in cancer onset and progression.87,88
Since unconjugated BAs are produced by intestinal microbiota, the direct negative effects are mainly due to the high concentrations reached in the gastrointestinal lu-men.27,39,82,83 For example, duodeno-gastro-oesophageal reflux of BA might play a cancer-promoting role both in the stomach84,89,90 and in the oesophagus,91,92 and local pH is involved in this process.
BAs act also as signaling molecules involved in a number of systemic processes,93,94 including metabolism and tumorigenesis.88 As previously mentioned, the two main receptors are the FXR and the GPBAR-1. FXR is considered the intracellular sensor of BAs, is mainly expressed in the entero-hepatic system, and regulates the expression of genes involved in the control of BAs, lipid and glucose homeostasis95-97 as well as inflammatory process-es.95 FXR safeguards the maintenance of BA concentration within a physiological range to prevent BA accumulation and cellular damage.18,97 The extent of FXR activation varies with BA affinity: the primary CDCA is the strongest agonist, the secondary LCA (lithocholic acid), DCA BAs have lower activity, while the more hydrophilic BAs do not activate this nuclear receptor.18
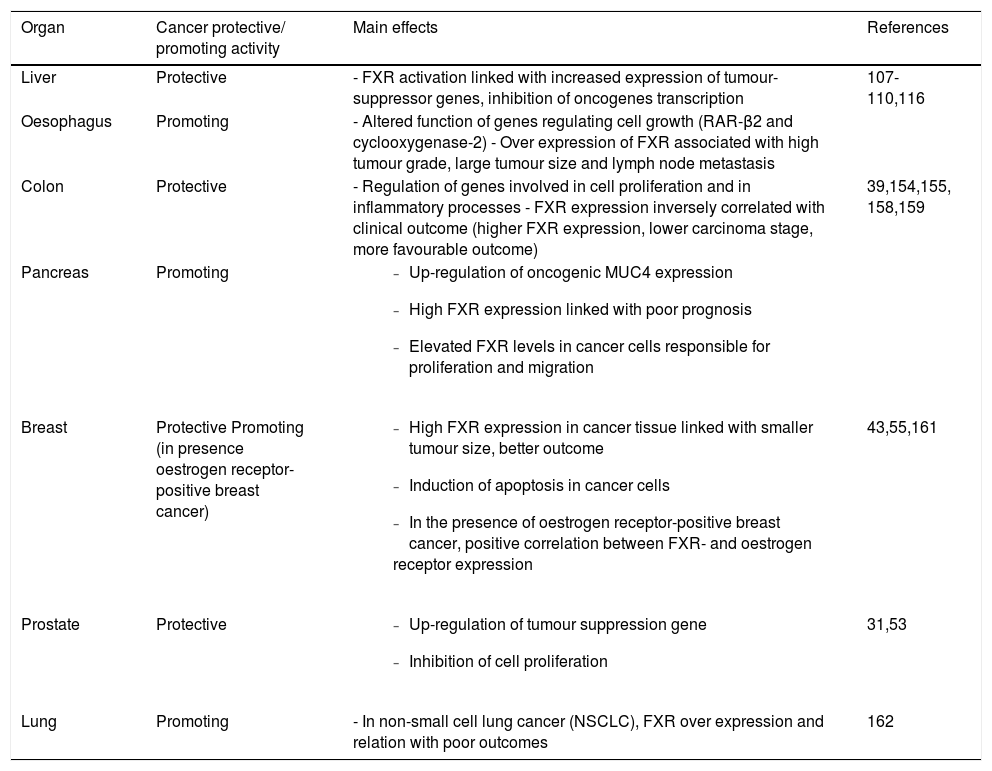
Of note, FXR is also able to govern the renewal of the intestinal epithelium and the regulation of proliferation of several cell types, including gastric,28 colon,29,39 oesopha-geal,40 pancreatic41,42 prostate,31 and breast43 cancer cells. FXR is expressed in several gastrointestinal and extra-intestinal organs,98 and the ultimate effect on promotion or inhibition of cancer onset/growth differs according to different anatomical sites (Table 1). Of course, this aspect merits additional studies.
Effects of FXR overexpression at the level of different anatomical sites.
| Organ | Cancer protective/ promoting activity | Main effects | References |
|---|---|---|---|
| Liver | Protective | - FXR activation linked with increased expression of tumour-suppressor genes, inhibition of oncogenes transcription | 107-110,116 |
| Oesophagus | Promoting | - Altered function of genes regulating cell growth (RAR-β2 and cyclooxygenase-2) - Over expression of FXR associated with high tumour grade, large tumour size and lymph node metastasis | |
| Colon | Protective | - Regulation of genes involved in cell proliferation and in inflammatory processes - FXR expression inversely correlated with clinical outcome (higher FXR expression, lower carcinoma stage, more favourable outcome) | 39,154,155, 158,159 |
| Pancreas | Promoting |
| |
| Breast | Protective Promoting (in presence oestrogen receptor-positive breast cancer) |
| 43,55,161 |
| Prostate | Protective |
| 31,53 |
| Lung | Promoting | - In non-small cell lung cancer (NSCLC), FXR over expression and relation with poor outcomes | 162 |
Hydrophobic BAs undergo continuous entero-hepatic re-circulation and can generate cell damage4,49 via a direct detergent cytolytic effect, increased hepatocyte apoptosis, neutrophil infiltration in the liver or combination of various factors.19 Altered microbiota, high-fat diet, involvement of liver and intestine might promote carcinogenesis by inflammation signaling.50,99 DCA promotes DNA damage and cellular senescence in hepatic stellate cells (senescence-associated secretory phenotype49), with initiation of inflammatory and tumour-promoting pathways potentially leading to liver cancer,48 in particular after exposure to chemical carcinogens.49 The secondary hydrophobic conjugated TCDCA showed a liver-cancer promoting activity in vitro in HepG2 cells: normal human liver cell proliferation increased significantly with down-regulation of the expression of a tumour suppressor gene (CEBPα), while in WRL-68 normal human hepatic cells, DCA, LCA and TCDCA upregulated the expression of oncoprotein c-myc. Furthermore, collaborative effects of a number of more hydrophobic BAs were able to promote liver can-cerogenesis in the mice undergoing nonalcoholic steato-hepatitis (NASH)/Hepatocellural carcinoma (HCC) changes after treatment with streptozotocin plus high-fat diet or high-fat diet alone.50 The emerging problem of non-alcoholic fatty liver disease, as a potentially evolutionary cause of liver disease worldwide leading to the necro-inflammatory NASH, progressive fibrosis, liver cirrhosis and HCC, needs to be also considered.100-105 Indeed, total fasting and post-prandial serum BAs are increased in patients with NASH compared to patients with healthy livers,106 suggesting a shift in BA composition (increased in taurine- and glycine-conjugated BAs and increased secondary BAs with sustained exposure to BAs possibly mediating liver injury). Thus, therapeutic strategies targeting microbiota, intestine and BAs retention and citotoxicity might indeed play a role in patients with obesity and non-alcoholic steato-hepatitis (NASH) exposed to long-term risk of liver cirrhosis and hepatocellular carci-noma.49,50
The BA-FXR-GPBAR-1 axis needs to be considered within the overall framework of liver tumorigenesis. FXR in the liver acts as a protective factor against cancer due to its role in maintaining BAs, glucose and lipid homeostasis, to its restoring capacity after liver injuries, to the ability of promoting hepatocyte protection and enhancing cell survival, to anti-inflammatory properties and to be a favourable gene-expression modulator (increase in expression of tumour-suppressor genes, inhibition of oncogenes tran-scription).107 CDCA and the synthetic FXR agonist GW4064 increase the expression of a tumour suppressor gene, NDRG2 (N-Myc downstream regulated gene 2), in human hepatoma cells and in primary hepatocytes. This property is abolished in FXR-knockdown animals and is increased with FXR over expression.108 The positive effects linked with FXR expression, however, are counterbalanced in the liver by a decreased FXR expression during processes leading to cancer onset.107 FXR-/- mice spontaneously develop (15 months of age) hepatocellular adenoma and carcinoma, with previous (9 to 12 months) liver injury and inflammation. Also in this case, an altered regulation of gene involved in the control of BAs levels is present, with high BAs concentration in both serum and liver. In this animal model, the role of endogenous BAs in cancer promotion appears evident, since administration of 2% cholestyramine is able to significantly reduce cancer lesions.109
Of note, a decreased FXR expression per se does not appear to be able, alone, to promote liver cancer onset and to maintain cancer proliferation if not associated with high levels of BAs. While the FXR deficiency may have a role as cancer promoter, an increment in BA levels is required for the promotion of cell proliferation and cancer forma-tion.110 Prospective metabolomics analysis of hepatocellu-lar carcinoma have clearly identified long term elevated serum BAs levels as a risk factor for cancer develop-ment.111 Additionally, mice with hepatocyte-specific FXR deficiency (FXR(hep-/-)) did not show spontaneous liver cancer formation with aging, but cell proliferation and cancer formation were induced by cholic acid supplementation by diet, and were linked with increased basal expression of tumour suppressor p53 protein and disturbance of the mitogen-activated protein kinases (MAPK) and JAK/STAT3 signaling pathways.110 The MAPKs signaling pathways, in particular, have a pathogenic role in a series of human diseases (including cancer) and their activation is secondary to cellular stress (also involving oxidative damage promoted by Bas112) and to the presence of proinflammatory cytokines.113 The activation of STAT3, on the other hand, is able to increase transcription of genes involved in suppression of anti-tumour immuni-ty,114 liver inflammation and cancer.114,115
An interesting animal model of FXR-null mice with re-expression of constitutively active FXR in enterocytes has recently suggested that, in the presence of reduced hepatic FXR expression, the reactivation of intestinal FXR normalized BA enterohepatic circulation through the fi-broblast growth factor 15 (FGF15)/cholesterol-7alpha-hy-droxylase enterohepatic axis, reducing BAs synthesis by the liver, with a protective effect from spontaneous HCC onset.116 Thus, in the case of reduced hepatic FXR expression, the coexistence of adequate entero-hepatic signaling pathways involving the FGF15/cholesterol-7alpha-hy-droxylase axis might be protective for liver cancer onset.
The role of aberrant signaling involving fibroblast growth factor 15/19, FGF receptor 4 (FGFR4) and beta-Klotho (KLB) co-receptor signaling system has been recently underlined in the onset of liver cancer,117 and altered pathways involving these additional key regulators of BA synthesis and metabolism are able to promote HC-Cin mice and to influence the clinical outcome in HCC patients.118
In mice, increased expression of FGF19 (fibroblast growth factor 19) promotes HCC development with FGFR4-dependent mechanisms and activating, also in this case, the STAT3 pathway.117 Higher concentrations of BAs (e.g. CDCA) might also explain in part the increased risk in men with Primary Biliary Cholangitis (PBC) for HCC, in particular in non-responders to UDCA therapy.119 FXR and the CDCA-dependent activation in the liver and intestine is likely involved.120,121
The GPBAR-1 receptor has also a key function in BA homeostasis, LCA and taurolithocholic acid (TLCA) being their most potent endogenous ligands.122-124 A BAs-stimulated GPBAR-1 expression is present Kupfer cells.125 Both FXR and GPBAR-1, once activated by BAs might lead to suppression of NF-κB factor and proinflammatory cytokines in the liver.126
Oesophageal cancerBarrett’s oesophagus is characterized by the development of metaplastic columnar epithelium that replaces the normal stratified squamous epithelium found in the distal oesophagus. Chronic gastroesophageal reflux disease (GERD) is the cause for Barrett’s oesophagus, which is a condition predisposing to the development of adenocarci-noma of the oesophagus.
In tissues from human Barrett’s oesophagus, DCA generated oxidative stress by inducing reactive oxygen and nitrogen species after acting on intracellular NADPH oxidase and mitochondria and activation of the NF-κB pathway.74,77,78,127 Cells hosting the damaged DNA might resist apoptosis.77 The BAs and acid-induced NF-κB activation in epithelial cells is dose- and time dependent and also involves the induction of COX-2 promoter activity, potentially contributing to the onset of oesophageal can-cer.78,128 By contrast, the more hydrophilic UDCA (urso-deoxycholic acid) protects from DNA damage and NF-κB activation.74,77,78 In a comprehensive study in patients with Barrett’s oesophagus, Peng, et al.74 showed that oral treatment with UDCA prevented the toxicity by DCA 250 μM (DNA damage, NF-κB activation in the metaplastic mu-cosa of patients with Barrett’s oesophagus). In vitro, UDCA activated the NF-E2-related factor 2 (Nrf2) to upregulate the expression of glutathione peroxidase 1 (GPX1) and catalase antioxidants, a finding further confirmed in biopsy specimens of Barrett’s metaplasia taken from patients after 8 week treatment with oral UDCA. The DNA-damaging effect might be operating with both glyco-conjugated BAs at acidic pH (pH = 4) but also with unconjugated BAs at higher pH (pH = 6). An overview on the role of secondary BAs in neoplastic development in the oesophagus is available by Cronin, et al.91
FXR might play a role also in the context of Barrett’s oesophagus: in the experimental mice model of oesopha-geal adenocarcinoma, the overexpression of FXR has been associated with higher tumour grade, larger tumour size and lymph node metastasis, and knockdown of FXR expression suppressed tumour cell growth. Results from this study indicated that FXR expression mediated BAs-induced alterations of genes regulating cell growth (RAR-β2 and cyclooxygenase-2).40
Gastric cancerWang, et al.47 studied gastric cancer in mice and found that acidified bile acids induce tumour progression and te-lomerase activity both in vivo and in vitro, with mechanisms involving higher c-Myc transcription (a regulator gene that codes for a transcription factor and is involved in cell cycle progression, apoptosis and cellular transformation), with increased expression of human telomerase reverse transcriptase (hTERT) at the protein and mRNA levels. In primary human gastric adenocarcinoma cancer cell lines MKN28, MGC803 and SGC7901, the same authors found that 100 μg DCA and CDCA under acidified media activate c-Myc that, in turn, increases hTERT expression.84 In the clinical setting, Tatsugami, et al.,90 studied 612 Japanese patients positive for H. pylori infection using gastric biopsies. The retrospective occurrence of gastric cancer was calculated in 357 patients followed by endoscopic examination for cancer screening for less than 3 years. BAs concentration in gastric juice correlated with the extent of gastric atrophy/intestinal metaplasia independently of inflammatory cell infiltration. Also, the occurrence of gastric cancer was increased in patients with high- as compared to those with low-BAs concentration. Exposure to acidified BAs (DCA and CDCA at pH 5.5) increased tumour progression in MGC803 gastric cancer cell line.
GPBAR-1 expression has been linked with advanced stages of gastric cancer; GPBAR-1 expression correlates with the expression of N-cadherin, a markers of epitheli-al-mesenchymal transition.129 Moderate to strong GPBAR-1 staining in gastric adenocarcinoma was associated with decreased patient survival, and BAs increased cell proliferation through activation of GPBAR-1 receptors and coupled G(q)α and Gα(i-3) proteins.46
Colon cancerColorectal cancer prevalence is dramatically rising worldwide.130 In the intestine, the replacement of intestinal villi cells is a crucial step. The process is completed every 3-5 days and starts from the pluripotent cells located at the bottom of intestinal crypts, which transform into specific enteroendocrine, absorptive, Gobleth and Paneth cells. From the top of the villi, apoptotic cells are released into the intestinal lumen at the end of the differentiation cycle. Several transcription factors are involved in these processes, namely the caudal-related homeobox transcription factor (CDX2), E-cadherin, claudin-2, genes like Mu-cin 2 and sucrose isomaltase. Further signaling pathways include Wnt/β-catenin, the cytoplasmic protein β-catenin and/or the tumour suppressor APC binding to β-catenin. For colorectal cancer onset, several mutations are required, starting from APC gene and also involving KRas, TP53, phosphoinositide 3-kinase (PI3K) and transforming growth factor β(TGFβ).39
Over-consumption of a Western-style diet can represent a step linking BAs to colorectal cancer. The Western-style diet brings excess calories, is enriched with highly-saturated fats and processed carbohydrates but lacks mono-polyunsaturated fatty acids and plant-derived proteins and fibre.34,39,83 Following Western-style/high-fat/ low-fibre diet, therefore, abnormally high levels of secondary BAs might increase in the intestine,131,132 and this step leads to disruption of the complex mechanisms governing the intestinal epithelial renewal. Elevated luminal concentrations of secondary DCA and LCA (at variance with the hydrophilic tertiary hydrophilic UDCA) might provoke intestinal cytotoxic damage which parallels the effect of other genetic and environmental factors acting as tumour promoter stepin the post-initiation early stages of colon carcinogenesis133 and acting as a tumour-promoting effect.134 Even cholecystectomy, a condition which increases the exposure of intestinal mucosa to elevated BA levels has been considered as a predisposing condition to colorectal cancer.135 Mechanisms of BA-induced tumori-genesis include DNA oxidative damage, hyperprolifera-tion, NF-κB activation and inflammation, β-catenin signaling and p53 degradation. Several additional mechanisms have been advocated and include BA-induced pro-liferative effect on undifferentiated epithelial cells of intestine136 and colon cells,137 disrupted colonic mucosal integrity,138 activation of extracellular signal-regulated ki-nase (ERK) signaling and epidermal growth factor receptor (EGFR)139 and stimulation of colonic epithelial proliferation via protein kinase C (PKC).140 Initiation of apoptosis resistance by BAs such as DCA and LCA141 would imply mitochondrial damage with mitochondrial oxidative stress, generation of reactive oxygen species (ROS), cytochrome C (cytC) release and activation of cy-tosolic caspases.71 Nuclear factor kappa β (NF-κB) pathway activation and release of arachidonic acid might work in concert with cytotoxic BAs in the colon.142
The intestinal microbiota is another important player in the scenario mentioned above. Microbes populate the human gut reaching massive concentration in the colon (up to 1012 CFU/g luminal content),143 play a key role in BA biotransformation from primary to secondary molecules and can be easily modulated by factors like age, nutrition, diseases, drugs and/or intestinal anatomy.144-146 Diet can heavily influence the microbial metabolic pathways and gas production,143-147 since the saccharolytic fermentation of carbohydrates by microbiota produces short-chain fatty acids (SCFAs) such as butyrate, propion-ate, acetate, and butyrate has anti-inflammatory and antine-oplastic properties148-150 while a high-fat diet would activate pathways involving proteolysis, inflammation and tumorigenesis.151,152 Zeng, et al.83 demonstrated that bu-tyrate (the short-chain fatty acid and microbiota-depend-ent metabolite of dietary fibre) at a concentration of 0.5-2.0 mM counteracted the detrimental effects of DCA (0.05-0.3 mM) on colon cell proliferation. Although both butyrate and DCA inhibited cell proliferation and increased cell ap-optosis rate, only butyrate increased G1 and G2 fractions (vs. only G1 with DCA) with a concomitant drop in the S-phase fraction at cell cycle analyses. DCA but not butyrate increased intracellular pathways including reactive oxygen species, genomic DNA breakage and the activation of ERK1/2, caspase-3 and PARP. Overall, the current data suggest that both butyrate and DCA inhibit colonic cell proliferation. However, butyrate increases tumour suppressor gene expression, whereas DCA decreases tumour suppressor activation in cell cycle and apoptosis path-ways.83 Similar mechanisms have been described in normal and tumour human colon cells,34 as well as in the mice model of colon cancer,24 where DCA and CDCA are able to cause oxidative DNA damage27,82 and apoptosis153 through oxidative processes which can be limited by the concomitant exposure of cells to antioxidants, i.e. beta-carotene, alpha-tocopherol, Na-butyrate, zinc and/or chlo-rogenic acid.24,27,82 Such findings point to a potential protective role, partly BA-mediated of healthy diets.
FXR expression has also a role in colon cancer,154,155 since mechanisms of cancerogenesis in the colon also involve Apc gene mutation, CDX2 inactivation and increased CpG methylation in the Fxr gene, resulting in loss of FXR in the colonic epithelium, increased mitotic activity, cell hyperproliferation; all features associated with a pro-tumorigenic phenotype.142,156,157 If FXR becomes deficient in the intestine, moreover, secondary BAs might be increased and less detoxified in the liver. Loss of FXR generates high BAs concentrations and, in animal models, a pro-tumorigenic phenotype39 with pathways similar to those observed for liver cancer. In an animal model, loss of FXR in the ApcMin/+ mice lead to early mortality and increased colon cancer progression, pointing to a protective role of FXR on intestinal cancer. However, the cancer-promoting effect was independent from intraluminal BAs, since it was not inhibited by treatment with cholesty-ramine.155 In mice, FXR deficiency also generates an up-regulation of genes involved in cell proliferation and in inflammatory processes, an increment in colon cell proliferation and a growth of small intestine adenocarcinomas in adenomatous polyposis coli mutant animals.158
In human colon cancer, FXR expression is repressed during the transition of adenoma to carcinoma and is not expressed in undifferentiated colon cancer cells SW480 and in metastasis derived SW620 cells.159 A systematic im-munohistochemistry mapping on human intestinal muco-sa showed that FXR expression was reduced in colon carcinomas as compared with non-neoplastic mucosa and that a relationship was evident between the loss of FXR expression and the grading of tumours in the right colon. FXR expression was inversely correlated with the clinical outcome of patients (higher FXR expression, lower carcinoma stage and more favourable outcome).154
Pancreatic cancerA relationship between BAs and pancreatic cancer has been suggested. BAs might reflux into the pancreatic duct and, on the other hand, are linked at a systemic level with obesity, diabetes and hypertriglyceridemia, all well known risk factors for pancreatic cancer.51 Elevated levels of BAs have been reported in serum and in pancreatic juice from patients with pancreatic cancer, as compared with controls. This finding might be linked to up-regulation of on-cogenic MUC4 expression.42 High expression of FXR in colon154 and breast43 cancer relates with better clinical outcome of patients. However, for pancreatic cancer, high FXR expression is rather linked with poor prognosis and poor survival. FXR elevation in pancreatic cancer cells might be responsible for cellular proliferation and migra-tion.41
Prostate cancerPositive effects of FXR overexpression have also been described in the case of prostate. FXR activity, in fact, is present in normal and cancer prostate epithelial cells and its stimulation by CDCA treatment is able to inhibit cell proliferation in prostate cancer.53 The suppression of prostate tumour growth is associated with decreased mRNA and protein levels of sterol regulatory element binding protein 1 (SREBP-1),53 and through an up-regulation of the tumour suppression gene for the Phosphatase and tensin homolog (PTEN) induced by the FXR overex-pression.31
Breast cancerFXR has been also detected in breast tissue.160 Similarly to that previously observed in colon cancer,154 in women with invasive breast carcinoma, high FXR expression in cancer tissue was linked with smaller tumour size and patients with high FXR expression had a better clinical outcome (longer overall and disease-free survival time) as compared with those with low FXR expres-sion.161In vitro, the activation of FXR by CDCA or by a synthetic ligand (GW4064) induced cell death (mainly by intrinsic apoptotic pathway) in four distinct phenotypes of breast cancer cell lines, without stimulating migration in cell lines.43 The effect of FXR overexpression on breast cancer, however, seems to be different (opposite) in the presence of oestrogen receptor-positive breast cancer, where a positive correlation was found between FXR- and oestrogen receptor expression. In this case, increased FXR levels were also correlated with the proliferation marker Ki-67 and nodal metastasis in postmenopausal women. The proliferation of oestrogen receptor-positive breast cancer could be, in this case, secondary to a crosstalk between FXR and oestrogen receptors, in particular during oestrogen deprivation (i.e. post-menopausal women, therapy with aromatase inhibi-tors).55
Lung cancerA recent study has also depicted a negative role of FXR expression in non-small cell lung cancer (NSCLC). In this case FXR is overexpressed and is related with poor outcomes in patients, in particular in the presence of concomitant over expression of cyclin D1,162 increment in Cyclin D1 protein and mRNA expression.163
Epigenetic FactorsThe pathway linking BAs and nuclear receptors with cancer onset is influenced by changes in gene expres-sion.42,47,50,83,84,95-97,107,164 This step leads to both benign and malignant diseases and is also able to influence the clinical outcome in cancer patients.118,154,161,162
The expression of genes involved in BAs-dependent signaling processes may be silenced, reduced or amplified by epigenetic mechanisms (mainly microRNA expression, DNA methylation, histone/gene acetylation165) also induced by dietary habits164 and various environmental factors, without changes in DNA sequence.
MicroRNAMicroRNAs represent a class of small noncoding RNAs. They play a key role in a number of diseases (including human carcinogenesis) mainly through a down-regulation of various target genes.
MicroRNA-22 (miR-22) has a pronounced tumour-suppressive role in different organs166,167 including co-lon168 and liver cancer.169,170 The process is regulated by FXR expression in liver and colon.35 CDCA, due to its high affinity for FXR,18 increases miR-22 levels in liver and colon cells with a silencing effect on cyclin A2 (CCNA2). In FXR-knockout mice low miR-22 levels are associated with increased number of Ki-67-positive cells in the colon and in the liver. In humans, levels of miR-22 and CCNA2 are inversely correlated with colon and liver cancers.35 Human oesophageal adenocarcinoma samples display increased levels of miRNA 221 and miRNA 222, as compared with Barrett’s oesophagus samples taken from the same patients.36 Also, levels of both miRNA-221 and 222 in cultured cells were related with FXR activity in response to BAs exposure and inhibited mRNA translation of p27Kip1, promoting degradation of the transcription factor CDX2.36 It has to be underlined that altered expression p27kip1 leads to deregulated cell growth/differentiation, promoting the development of a number of tumours in humans.171
DNA methylationIn the rats and the mice, BAs like DCA, CDCA, CA and LCA introduced by diet induced DNA hypomethyla-tion in the colon. This effect was not induced by administration of the more hydrophilic UDCA.172 Other studies clearly point to a relationship between DNA methylation and FXR expression. Mutations in the adenomatous poly-posis coli (APC) gene have been linked with the early development of colorectal cancer.37 Studies in APC deficient mice suggest that FXR expression is reduced; this silencing effect is mainly linked to CpG methylation of the Fxrα3/4 promoter.156 In the same study DCA lowered CpG methylation of FXR and induced FXR expression in human HCT-116 but not HT-29 colon cancer cells.156 The relationship between DNA methylation and FXR silencing was also described in a previous study in human colon cancer, demonstrating a reduced expression/function of FXR in precancerous lesions and a silenced FXR in the majority of stage I-IV tumours.30 BAs are also able to affect DNA methylation in human oesophageal tissue. Exposure of human oesophageal epithelial cells to a mixture of six different forms of BAs (GCA, TCA, GCDCA, TCDCA, GDCA, and TDCA) induced Caudal-related homeobox 2 (Cdx2) expression (as an early marker of Barrett’s oesophagus) through promoter demethylation. This mechanism contributes to the onset of intestinal metaplasia, a premalignant lesion of oesophageal adenocarcino-ma.38 Over expression of Cdx2 was also described in human oesophageal tissues, in esophagitis and, in higher proportion, in samples from patients with Barrett’s oesophagus and primary oesophageal adenocarcinoma.45
Histone acetylation and chromatin remodelingPost-translational modifications of histones (i.e. his-tone acetylation/deacetylation) and chromatin remodeling are well-known epigenetic mechanisms173,174 working with transcriptional cofactors (i.e. sensing activities and signaling pathways,175 as FXR176) and have a defined role in the metabolism of lipids177 and in BA homeostasis and functions.178 The small heterodimer partner (SHP, an orphan nuclear receptor) is an important epigenomic regulator of BA biosynthesis, mainly acting through chromatin remodeling179,180 and histone deacetylation.181,182 SHP has been identified as having an antitumor role in liver can-cer183,184 due to its capacity to regulate cell proliferation, apoptosis, DNA methylation, and inflammation,184 and is also involved (due to its strict relationships with FXR) in colon,156 gastric185 and breast160 cancer.
In an animal model Sirtuin 1 (SIRT1), a key regulator of a number of metabolic processes (including BAs home-ostasis), has a critical role in the regulation of the regenerative response in the liver by post-trascriptional modifications involving FXR activity (through the acetylation of FXR and neighboring histones) and mTOR, potentially contributing to liver cancer onset through dys-regulation of BA homeostasis by persistent FXR deacetylation.181
Bas, Microbiota, Environmental PollutantsBAs undergo biotranformation especially in the colon, due to unique microbial enzymes which are encoded within the gut microbioma.1 Distribution of BSH enzymes, essential in primary conjugated BA deconjugation in the colon, are found in Gram positive species Lactobacil-lus, Enterococcus, Clostridium spp, gram negative Bacteroides spp and in several bacterial strains (i.e., L. plantarum, L. acido-philus, L. salivarius, C. perfringens, etc.). BSH in bacteria might confer a defensive mechanism against the effect of BAs and provide glycine and taurine as bacterial energetic source (glycine → NH4+CO2 and taurine → NH4+CO2+sulphate).1 Current knowledge suggests that BSH influences several physiological processes in the host and mark the BA signature with a control on metabolic, immunological, and receptorial functions.1,13 Further steps after bacterial deconjugation in the colon include anaerobic bacterial re-amidation, redox reactions, desulfation186 (as prevention of BA loss in feces/urines), esterification, oligomerization from time-to-time by Lactobacillus, Bacteroidetes, Eubacteria, Clostridium, etc.4,187,188 Bacterial stereospecific hydroxysteroid dehydrogenases (HSDH) control BA oxidation, epimerization and dehy-droxylation189 and, via Clostridium species, the biosynthesis of the tertiary UDCA from the secondary CDCA.190 Several other bacterial species will join such complex biosyn-thetic pathways.
Events pointing to qualitative or quantitative changes of intestinal microbial community may heavily influence bacterial enzymes and, in turn, BA composition and functions. Paradigmatic situations include germ-free or antibiotic treated animals,191,192 food consumption193,194 with changes occurring even in the short-term (1 to 3 days195), aging,196 inflammatory bowel disease,186 even metabolic disorders,197,198 functional disorders including irritable bowel syndrome,143,199 intestinal surgery including bariat-ric surgery in morbid obesity,200,201 primary sclerosing cholangitis202 and ingestion of environmental toxics contained in water or food.66-69,203-209
Forms of intestinal dysbiosis might also contribute to tumorigenesis in different ways. Obesity is a major risk factor for several types of common cancer,210 and obesity might induce changes in gut microbiota,211 shift the BA pool profile (i.e. increased DCA), and several hydropho-bic BAs might collaboratively promote carcinogenesis (not HCC initiation) via DNA damage,4 induction of senescence-associated secretory phenotype (SASP) in hepatic stellate cells (HSCs),49 Gram-negative activation of toll-like receptor (TLR) 4 and bacterial production of li-popolysaccharide (LPS) in the intestine.212 In mice, prevention of liver cancerogenesis has been achieved by blocking DCA formation, and acting on gut microbiota49,50 with sterilization,212 increasing intestinal excretion of hy-drophobic BAs (i.e. with the bile acid sequestrant choles-tyramine50). Similar mechanisms involving disrupted BA pool and dysbiosis might also operate in other sites of human tumorigenesis. In the colon DCA and LCA would act as procarcinogenic bacterial metabolites but also promising therapeutic targets.213 Both BAs might act as proin-flammatory agents, eliciting the production of reactive oxygen and nitrogen species, as well as NF-κB activation in intestinal epithelial cells.214-217 Moreover, chronic exposure to DCA induces the production of DNA adducts which parallels enhanced epithelial cell proliferation and decreased apoptosis.34
Tumorigenesis can also imply an impaired interaction between BAs and their receptors.14 FXR, for example prevents excessive inflammation in the liver and intestine218 (see also previous paragraphs on BAs and FXR). Thus, while changes in microbiota might be implicated in some steps of tumorigenesis, inducible changes of microbiota might also represent an additional clue to cancer thera-py.219,220 Much caution, however, is required in this field, until definitive prospective clinical/population studies will clarify the true pathogenic role of this consortium of actors in carcinogenesis.
Recent studies point to the marked effects on intestinal microbiota of some environmental pollutants as heavy metals (mainly arsenic, cadmium and lead) and persistent organic pollutantsingested with contaminated water or food,66,203-206 resulting in an increased toxicity (and potential mutagenic properties) of the BAs pool. This induces oxidative stress221 and strongly alters the intestinal micro-biota, by reducing the amount of both primary and secondary BAs. This mechanism develops through a down-regulation of CA, UDCA and DCA levels.203 A marked alteration of gut microbiota has been reported in the animal model, after ingestion of arsenic in drinking water, which also increased the excretion of 7-α-hydroxy-3-oxo-4-cholestenoate (involved in the biosynthesis of primary BAs) and reduced GCA in fecal samples of treated animals.66 Of note, 7-α-hydroxy-3-oxo-4-cholestenoate is believed to be, in humans, an important precursor of CDCA,222 the strongest agonist involved in FXR activa-tion,18 and GCA has been linked by metabolomics with hepatocellular carcinoma.223,224
Pesticides such as chlorpyrifos,207-209 diazinon,67 and 2,3,7,8-tetrachlorodibenzofuran (TCDF)69 can greatly alter microbiota composition58,157-159 (Figure 1).

Diazinon, a widely employed organophosphate pesticide able to contaminate ground water, drinking water wells and food, in an animal study strongly altered gut microbiota and the related metabolic functions with different sex-specific patterns (more pronounced responses in male mice). Significant increments in Bacteroidaceae Bacteroides (> 2,000-fold rise, bacteria with bile salt hydro-lase enzymes, BSHs) and Proteobacteria (+15-fold rise, bacteria involved in BA transformation) were recorded in treated animal. As a consequence of this increased decon-jugation potential, a 4-fold and a 5-fold increment in LCA levels was recorded in treated male and female mice, respectively and, in female mice, a significant increment (3.6-fold) of DCA was also noticed.67
Chlorpyrifos is an organophosphate pesticide which acts on the nervous system by inhibiting acetylcholineste-rase. This compound promoted alterations in gut micro-biota composition and metabolome (including alterations of the BAs pool) in mice. Changes were associated with histological modifications in the colon of treated animals, intestinal inflammation and altered permeability.209 In other animal models, chronic exposure to chlorpyrifos at low doses caused intestinal dysbiosis with proliferation (Ente-rococcus and Bacteroides) or decrement (lactic acid bacteria as Lactobacillus and the bifidobacteria) of selected strains.207
Oral exposure of female rats during gestation to the same pesticide caused marked gut dysbiosis and damages to the intestinal epithelium in the pups.208
The SHIME® model also demonstrated that chlorpyrifos is able to affect human colonic microbiota, with an increase in Enterobacteria, Bacteroides and Clostridia, and a decrease in bifidobacterial counts following chronic low (below-threshold) doses of CPF (1 mg/day for one month, dissolved in rapeseed oil).68
Similar results were promoted by 2,3,7,8-tetrachlorod-ibenzofuran (TCDF), a persistent organic pollutant potentially introduced with diet. TCDF in mice markedly altered gut microbiota by shifting the ratio of Firmicutes to Bacteroidetes; this change was associated with increased levels of DCA in the small intestine and feces, inhibited the FXR signaling pathway (i.e. down-regulation of FXR mRNA and its target gene small heterodimer partner [SHP] mRNA) in both the ileum and liver.69
Potential Cancer Promoting Effects from Interactions Between Alcohol, Smoking, and Ba HomeostasisIncreased risk of cancer can also partly result from the influence of lifestyle on BA homeostasis. Alcohol consumption and smoking, in particular, are well known risk factors for gastrointestinal cancers225,226 and have specific relationships with BAs metabolism.
Alcohol ingestionAcute ethanol ingestion generates a dose-dependent increment in the biosynthesis of BAs in humans with in situ gallbladder,60 and alcohol abuse has been linked with increased fecal BA excretion.59 Alcohol can significantly alter hepatic BAs homeostasis through modulation of intestinal microbiota227 and increasing BAs synthesis through an increased gene expression and activation of Cyclic AMP responsive element binding protein, hepatic specific (CREBH),63 an endoplasmic reticulum-tethered transcription factor known to be a key factor in the regulation of hepatic lipid homeostasis. A down-regulation of FXR by alcohol has been described, with a consequent increase in BAs synthesis and hepatic BA pool.228,229 Furthermore, in rat, chronic alcohol ingestion lead to marked variations of the BAs pool, with a reduction in taurine-conjugated BAs and a rise in glycine-conjugated BAs (more toxic) at the level of liver and in the gastrointestinal tract (duodenum and ileum).229
Chronic alcohol ingestion is also able to strongly affect the entero-hepatic circulation of BAs through well documented effects on BAs transporters both in the liver228,229 and in the ileum,229 finally leading to increased serum levels BAs.
Cigarette smokingSmokers show altered gut microbiota,230 increased BAs reflux in the stomach and increased intra-gastric bile salts concentration.231 Moreover nicotine, a primary component of cigarette smoking, is able to enhance the oxidative capacity of sodium DCA, increasing its genotoxic properties.64
In an animal model, the coexistence of gastro-oesopha-geal reflux of BAs and cigarette smoking aggravates the onset of Barrett’s oesophagus and potentially accelerates the progression to oesophageal cancer through a strong induction of cyclooxygenase-2 (COX-2) expression and a 10-fold increase in 4-aminobiphenyl (4-ABP) protein adducts.65 Increased expression of FXR in human small airway epithelium with staining scores negatively correlated with FEV 1% predicted of smokers without and with chronic obstructive pulmonary disease. The correlation also existed with CDCA leading to increase in COX-2 expression in bronchial epithelial cells. In the same study, FXR expression was induced by IL-4 and IL-13 in human bronchial epithelial cells and by exposure to cigarette smoke in rats.232
ConclusionsBAs are key regulators of complex homeostatic mechanisms at a systemic level ranging from cell proliferation to modulation of inflammation, interaction with the family of nuclear receptors, immunity and metabolic processes. Several pathways can be disrupted and predispose to cancer onset and progression in digestive and extra-digestive organs (Figure 2). The nuclear receptor FXR, in this respect, acts as a major sensor of BA in the liver and in the intestine and is deemed as a tool able to prevent excessive inflammation.14 Several evidences point to a key role for BA-FXR also in tumorigenesis. Proinflammatory factors are over expressed in the liver and colon of FXR-null mice, namely interleukin-6, interferon y, Tumor Necrosis Factor-cc,125,158 and NF-kB is leading chronic inflammatory changes in both liver and intestine,233 and is inhibited in vitro by FXR activation with GW4064.234,235 Also, FXR-null mice develop spontaneous liver cancer11,109 while hepatocellular carcinoma might be a late complication of the inflammatory non-alcoholic steato-hepatitis (NASH).19,100 BAs administered exogenously promote tumorigenesis in the liver either in mice and rat model.109,236,237 In the clinical setting, children with progressive familial intrahepatic cholestasis type 2 (PFIC type 2) have increased prevalence of hepatocellular carcinoma in a background of elevated plasma and intrahepatic BA concentrations.238
. PXR: Pregnane X receptor. VDR: Vitamin D receptor.")
Furthermore, pathways involving the intestinal micro-biota and epigenetic factors regulating gene expression act as a common interface between environmental factors (including diet, lifestyle, exposure to environmental toxics) and the molecular events promoting the onset and the progress of cancer. The high-fat diet, for example, increases the fecal concentration of secondary BAs and is a risk factor for the development of colorectal cancer.
Of note, intestinal microbiota and the epigenome are modifiable factors and, thus, might be modulated by both primary prevention strategies (i.e. changes in dietary habits and lifestyle, reduced exposure to environmental toxics) and therapeutic tools. Future studies are needed to better clarify how these measures could influence pathogenic mechanisms leading to disease onset and progression and if they will also be able to ameliorate the efficacy of the available therapeutic tools.
On the other hand, the therapeutic role of hydrophilic BAs (mainly UDCA, TDCA) counterbalancing the direct (cytotoxicity) and indirect (mainly in term of gene expression and activity of nuclear receptors) negative effects of the more hydrophobic BAs needs to be more clearly assessed in both digestive and extra-digestive cancers.
Abbreviations- •
AQPs: aquaporins.
- •
BAs: bile acids.
- •
CDCA: chenodeoxycholic acid.
- •
DCA: deoxycholic acid.
- •
FGF15: fibroblast growth factor 15.
- •
FGF19: fibroblast growth factor 19.
- •
FGFR4: FGF receptor 4.
- •
FXR: farnesoid X receptor.
- •
GPBAR-1: G-protein-coupled bile acid receptor-1 (also known as TGR5).
- •
LCA: lithocholic acid.
- •
UDCA: ursodeoxycholic acid.
We declare that we have no conflicts of interest.
AcknowledgementsThe present chapter is written in the context of the project FOIE GRAS, which has received funding from the European Union’s Horizon 2020 Research and Innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 722619. Emilio Molina-Molina and Raquel Lunardi Baccetto are recipients of Foie Gras Early Research Training Grant.



