











Hepatitis C is a common cause of end-stage liver disease, and the main indication for liver transplantation in Latin America. Treatment of hepatitis C infected patients improves important long-term outcomes as mortality. Sustained viral response is reached in near 50% of patients with the previous management based in pegylated interferon and ribavirin. Recently new drugs were available increasing sustained viral response significantly, changing the standard of care to triple therapy. This guidelines provides a framework for practitioner in Latin America, to the management of patients with hepatitis C chronic infection.
Recent advances in the treatment of hepatitis C virus (HCV) infection have been published. Recent phase III trials of the first-generation protease inhibitors (PIs) boceprevir (BOC) and telaprevir (TVR) open a new page in the therapeutic paradigm for HCV infection. These new drugs also bring another perspective for the assessment, management, and complications related to the therapy.
This update on the management of chronic HCV infection aims give to the practitioners in Latin American countries an evidence-based framework for assessing, treating, and managing complications in HCV-infected patients. The main objective of this review is to set the guidelines for the use of PIs in the treatment of chronic HCV-infected patients who are either naïve or nonresponders to a previous course of pegylated interferon/ribavirin (PR).
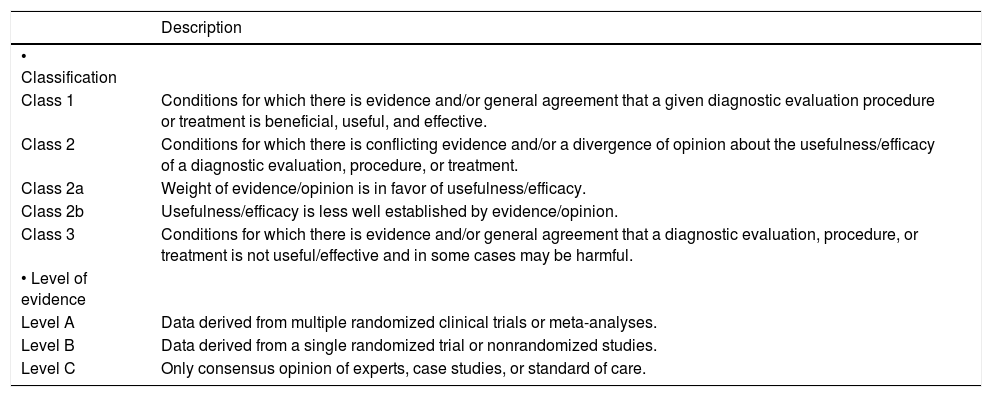
This information is reported within the framework of the recommendations for the Standardized Reporting of Clinical Practice Guidelines1 (Table 1).
Grading system for recommendations.
| Description | |
|---|---|
| • Classification | |
| Class 1 | Conditions for which there is evidence and/or general agreement that a given diagnostic evaluation procedure or treatment is beneficial, useful, and effective. |
| Class 2 | Conditions for which there is conflicting evidence and/or a divergence of opinion about the usefulness/efficacy of a diagnostic evaluation, procedure, or treatment. |
| Class 2a | Weight of evidence/opinion is in favor of usefulness/efficacy. |
| Class 2b | Usefulness/efficacy is less well established by evidence/opinion. |
| Class 3 | Conditions for which there is evidence and/or general agreement that a diagnostic evaluation, procedure, or treatment is not useful/effective and in some cases may be harmful. |
| • Level of evidence | |
| Level A | Data derived from multiple randomized clinical trials or meta-analyses. |
| Level B | Data derived from a single randomized trial or nonrandomized studies. |
| Level C | Only consensus opinion of experts, case studies, or standard of care. |
There are three main scenarios in which laboratory tests for monitoring HCV infection are important for management:
- •
Pretreatment evaluation.
- •
Monitoring the response to treatment.
- •
Follow-up of noncandidates for treatment.
IL28B (interleukin-28B) genotype testing was used originally as a major predictor of the response to PR therapy. However, it remains a useful tool in the therapy decision-making process in the era of direct-acting antiviral (DAA) agents. Beyond the initial use of IL28B genotype testing, it important to bear in mind that the IL28B CC genotype is more than twice as frequent in patients who clear HCV infection spontaneously compared with those who progress to chronic liver disease. The IL28B genotype provides information helpful for deciding whether to use a DAA-free regimen or to shorten therapy. The predictive value of IL28B genotype testing for a sustained virological response (SVR) is superior to that of the pretreatment HCV RNA load, fibrosis stage, age, and sex, and is higher in patients infectled by the HCV genotype 1 (GT1) virus than in those infected by the GT2 or 3 virus.2,3
IL28B genotype testing has limited value for patients needing treatment because of advanced fibrosis or for those identified for treatment; for patients with mild disease for whom therapy could eventually be deferred, the finding of a CC genotype might help in deciding whether to initiate short-duration (i.e., 24-week) treatment.4
Beyond the original concept that IL28B genotype is associated with IFN responsiveness, recent data show that IL28B genotype may also predict the outcome in HCV GT1-infected patients treated with IFN-free regimens.5
- •
Recommendation: IL28B genotype testing should be considered whenever the physician or patient needs additional information for predicting more accurately the probability of achieving an SVR and for deciding on the most suitable therapy duration (Class 2a, Level B).
A low HCV viral load (< 800,000 IU/L) is associated with a higher PR treatment response. A low HCV viral load linked to a rapid virological response (RVR) is associated with a higher SVR using a PR DAA-free regimen. In these patients, therapy may also be shortened to 6 months. HCV viral load should always be tested when evaluating a patient for antiviral treatment.6 There is no current agreement on the most discriminatory HCV RNA level, which ranges from 400,000 to 800,000 IU/mL (5.6-5.9 log10 IU/mL).
Monitoring the Response to TherapyResults of HCV viral kinetics should be considered when deciding on the duration of antiviral treatment. In the PI era, physicians should identify both the liver disease stage (especially to detect cirrhosis) and whether pertinent information about the previous treatment response is available in treatment-experienced patients, who should be classified as relapsers, partial responders, or null responders. Patients initiating PR require measurement of the HCV RNA viral load at baseline, at weeks 4, 12, and 24 of treatment, at the end of treatment, and 24 weeks after the end of therapy.7
In all phase III studies of BOC and TVR, HCV RNA quantification was performed using the COBAS HCV RNA 2.0 TaqMan test. This technique has lower limits of HCV RNA detection and quantification of 25 IU/mL and 9.3 IU/mL, respectively.8,9 When monitoring therapy, the viral load results must be available on time to avoid incorrect decisions about treatment duration.
Clinical impact of monitoring HCV viral kinetics during treatment with BOC:
- •
Week 4. Strong predictive value, and mainly reveals IFN sensitivity.
- •
Weeks 8 and 24. Information for the suitability of response-guided therapy (RGT) according to an early and slow or delayed response.
- •
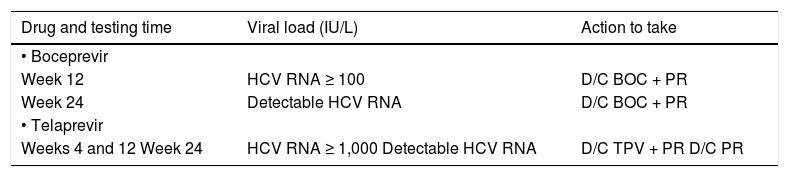
Weeks 12 and 24. Information about the lack of treatment efficacy (i.e., futility) as a basis for stopping rules10 (Table 2).
Table 2.Stopping rules for boceprevir and telaprevir.
Drug and testing time Viral load (IU/L) Action to take • Boceprevir Week 12 HCV RNA ≥ 100 D/C BOC + PR Week 24 Detectable HCV RNA D/C BOC + PR • Telaprevir Weeks 4 and 12 Week 24 HCV RNA ≥ 1,000 Detectable HCV RNA D/C TPV + PR D/C PR D/C: discontinuation. PR: pegylated interferon/ribavirin. BOC: boceprevir. TPV: telaprevir.
Clinical impact of monitoring HCV viral kinetics during treatment with TVR:
- •
Weeks 4 and 8. Information about RGT applicability according to the early and slow or delayed response (only for treatment-naïve patients and relapsers).
- •
Weeks 4 and 12. Information for stopping rules if HCV RNA levels are ≥ 1,000 IU/mL.
- •
Weeks 12 and 24. Information for stopping therapy with PR when HCV RNA is detected11 (Table 2).
Monitoring of noncandidate patients for therapy should assess mainly liver disease progression. Decreased platelet count, increased ratio of aspartate aminotransferase to alanine aminotransferase (AST/ ALT ratio), and prolonged prothrombin time are the earliest indicators of progressive liver fibrosis and portal hypertension. The frequency of the biochemical monitoring tests will depend on the patient’s age, stage of liver disease, and comorbid conditions.12 Emphasis should be placed on the careful selection and monitoring of patients. This requires professional expertise, and structured and multidisciplinary support for the proper management of adverse events (AEs). One of the most important objectives of HCV monitoring during treatment is to avoid exposing patients to AEs and variants that are resistant to the new DAAs that will be marketed in the future.
Liver Fibrosis Biomarkers in Patients With HCV InfectionThe prognosis and management of patients with chronic HCV infection depend on the amount and progression of the liver fibrosis and the risk of cirrhosis. Over the past decade, the role of the liver biopsy for staging of fibrosis has been challenged by the development of noninvasive methodologies.13 Liver biopsy is an imperfect gold standard; hence, in patients with HCV and in other patients, optional noninvasive methods have been explored for detecting and grading liver fibrosis. Liver biopsy analysis has several limitations; it is an invasive procedure that is prone to sampling errors and to intra- and interobserver variation.14 Severe complications such as hemoperitoneum, biliary peritonitis, and pneumothorax are rare and have been reported in 0.3–0.5% of cases. Death is exceedingly rare but has been reported occasionally in patients with advanced liver disease, a hemorrhagic tumor, or major comorbidities. A liver biopsy should be performed only after carefully balancing the risks of the procedure with the potential benefits in terms of patient management.15
Noninvasive MethodsCurrently available noninvasive tests rely on two different but complementary approaches: a biological approach based on serum biomarkers of fibrosis and a physical approach based on the measurement of liver stiffness by shock waves, ultrasound, or magnetic resonance elastography (Table 3). When using both approaches, it is important that the time between noninvasive tests is as short as possible because the results of liver function tests or other biological markers can change.
Noninvasive methods for diagnosing liver fibrosis.
| Serum biomarkers of HCV: |
| • FibroTest: formula combining α–2 macroglobulin, γGT, apolipoprotein A1, haptoglobin, total bilirubin, age, and sex. |
| • Forn’s index: 7.88–3.131 × In(platelet count) + 0.781 x In(GGT) + 3.467 × In(age) - 0.014 × (cholesterol). |
| • AST to platelet ratio (APRI) = (AST/upper limit of normal)/platelet count (expressed as platelets × 109/L) × 100. |
| • FIBROSpect II: formula combining α–2 macroglobulin, hyaluronate, and TIMP–1. |
| • MP3 = 0.5903 × log10 PIIINP (ng/mL) - 0.1749 × log10 MMP–1 (ng/mL). |
| • Enhanced liver fibrosis score (ELF): formula combining age, hyaluronate, MMP–3, and TIMP–1. |
| • Fibrosis probability index (FPI) = 10.929 + (1.827 × lnAST) + (0.081 × age) + (0.768 × past alcohol use) + (0.385 × HOMA-IR)-(0.447 × cholesterol). |
| • Hepascore: formula combining bilirubin, GGT, hyaluronate, α–2 macroglobulin, age, and sex. |
| • FibroMeter: formula combining platelet count, prothrombin index, AST, α2 macroglobulin, hyaluronate, urea, and age. |
| • Lok index = –5.56 - 0.0089 × platelet count (103/mm3) + 1.26 × (AST/ALT) + 5.27 × INR. |
| • Goteborg University Cirrhosis Index (GUCI) = AST × prothrombin - INR × 100/platelet count. |
| • FibroIndex = 1.738 - 0.064 × (platelet count [104/mm+]) + 0.005 × (AST HU/LI) + 0.463 × (a2-globulin [g/dL]). |
| • FIB–4 = age (years) × AST (IU/L)/(platelet count [109/L]) x (ALT [U/L])172. |
| • HALT-C model = –3.66 - 0.00995 × platelet count (103/mL) + 0.008 × serum × TIMP–1 + 1.42 × log(hyaluronate). |
| Measurement of liver stiffness: |
| • Transient elastography, FibroScan: results 2.5–75 kPa. |
| • Acoustic radiation force impulse imaging, ACUSON 2000 Virtual Touch Tissue Quantification: results 0.5–4.4 m/s. |
| • Magnetic resonance elastography: results 0.5–10 kPa. |
Key factors required for acceptance of a liver biomarker in clinical practice:
- •
Accuracy.
- •
Reproducibility.
- •
Liver specificity.
- •
Ability to predict stability, progression, and regression.
- •
Correlation with meaningful endpoints.
- •
Patient acceptability.
- •
Clinical and research use.
- •
Affordability.
Considering the wide availability of noninvasive methods in Latin America, there is no specific recommendation for one method, and the clinician should consider those that are available and affordable in the specific setting. At present, the liver biopsy plays an important role in Latin America.
Serum BiomarkersMany serum biomarkers have been evaluated for their ability to identify liver fibrosis. Direct markers that reflect the deposition or removal of extracellular matrix in the liver include serum levels of hyaluronate, laminin, YKL-40, procollagen III, N-peptide, type IV collagen, collagenases, metalloproteinases, and tissue inhibitory metalloproteinase 1. Indirect markers include factors that can be measured in routine blood tests such as the prothrombin index, platelet count, and AST/ ALT ratio.16
The practical advantages of analyzing serum biomarkers to measure fibrosis include their high applicability and interlaboratory reproducibility, their potential widespread availability and in some cases, the low cost of the aspartate to platelet ratio index (APRI). The disadvantages of these serum markers are their lack of specificity as indicators of liver fibrosis, inability to discriminate between intermediate stages of fibrosis, delay in the generation of results, high cost, and limited availability (proprietary restrictions) and use in some conditions (e.g., hemolysis, Gilbert syndrome, inflammation).17
Based on the results of a large-scale, multicenter study (n = 1,013) by Sebastiani, et al. in 2012, the sequential algorithm for fibrosis evaluation biopsy and the Fibropaca algorithm are attractive methods for large-scale screening of liver fibrosis in HCV patients in clinical practice. These algorithms may be particularly useful for screening HCV-infected individuals for whom an immediate approach involving a liver biopsy is problematic or questionable, such as elderly HCV carriers.18 In 2012, Castera, et al. reviewed the diagnostic performance of serum biomarkers of fibrosis for detecting significant fibrosis and cirrhosis. They found that, overall, biomarkers detect intermediate stages of fibrosis less accurately than they do cirrhosis; the most widely used and validated markers of fibrosis are the APRI and FibroTest.
Methods to Measure Liver StiffnessLiver fibrosis can be staged using one-dimensional transient elastography (TE), which measures the velocity of a low-frequency (50 Hz) elastic shear wave propagated through the liver. The velocity is directly related to tissue stiffness; i.e., the stiffer the tissue, the faster the shear wave propagates. The results are expressed in kPa and range from 2.5 to 75 kPa; a normal value is around 5 kPa.19
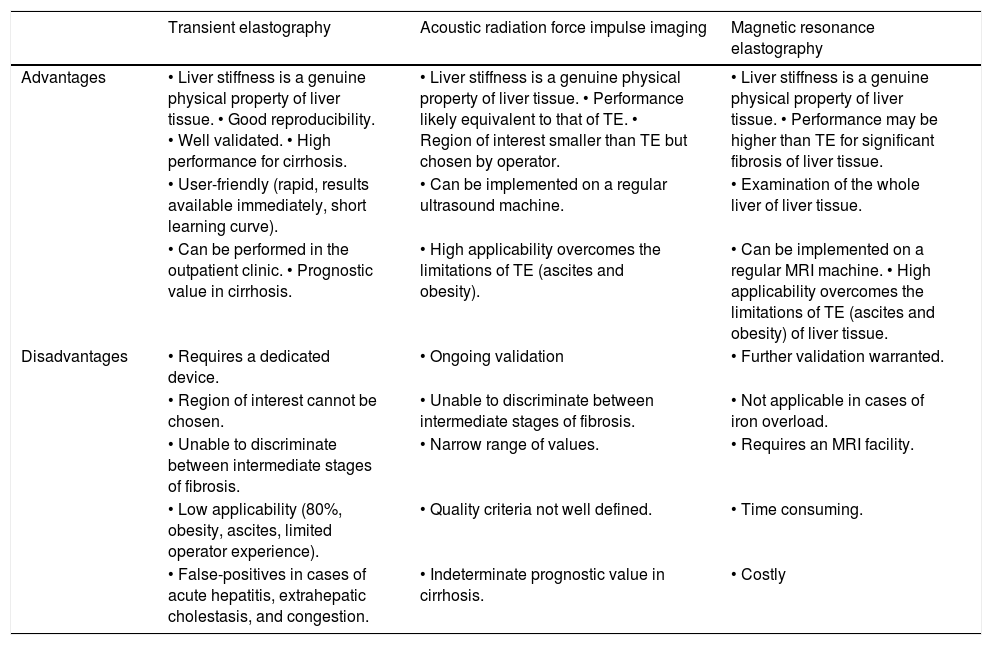
Several other liver elasticity-based imaging techniques are being developed, including two-dimensional acoustic radiation force impulse imaging, which involves mechanical excitation of tissue using short-duration (262 µs) acoustic pulses that propagate shear waves and generate localized, μ-scale displacements in tissue. The shear wave velocity (expressed in m/s) is measured in a smaller region that can be chosen by the operator. Three-dimensional magnetic resonance elastography uses a modified phase-contrast method to image the propagation characteristics of the shear wave in the liver.20 Elasticity is quantified by magnetic resonance elastography and is expressed in kPa using a formula that determinates the shear modulus, which is equivalent to one-third of the Young’s modulus used with TE.21 The advantages and disadvantages of methods for measuring liver stiffness are listed in table 4.17
Advantages and disadvantages of the methods to measure liver stiffness.
| Transient elastography | Acoustic radiation force impulse imaging | Magnetic resonance elastography | |
|---|---|---|---|
| Advantages | • Liver stiffness is a genuine physical property of liver tissue. • Good reproducibility. • Well validated. • High performance for cirrhosis. | • Liver stiffness is a genuine physical property of liver tissue. • Performance likely equivalent to that of TE. • Region of interest smaller than TE but chosen by operator. | • Liver stiffness is a genuine physical property of liver tissue. • Performance may be higher than TE for significant fibrosis of liver tissue. |
| • User-friendly (rapid, results available immediately, short learning curve). | • Can be implemented on a regular ultrasound machine. | • Examination of the whole liver of liver tissue. | |
| • Can be performed in the outpatient clinic. • Prognostic value in cirrhosis. | • High applicability overcomes the limitations of TE (ascites and obesity). | • Can be implemented on a regular MRI machine. • High applicability overcomes the limitations of TE (ascites and obesity) of liver tissue. | |
| Disadvantages | • Requires a dedicated device. | • Ongoing validation | • Further validation warranted. |
| • Region of interest cannot be chosen. | • Unable to discriminate between intermediate stages of fibrosis. | • Not applicable in cases of iron overload. | |
| • Unable to discriminate between intermediate stages of fibrosis. | • Narrow range of values. | • Requires an MRI facility. | |
| • Low applicability (80%, obesity, ascites, limited operator experience). | • Quality criteria not well defined. | • Time consuming. | |
| • False-positives in cases of acute hepatitis, extrahepatic cholestasis, and congestion. | • Indeterminate prognostic value in cirrhosis. | • Costly |
To increase the diagnostic accuracy of these tests, the sequential combination of biomarkers or the concomitant use of TE and biomarkers has been proposed. The combination of these strategies may be effective for the diagnosis of significant fibrosis and may lead to a reduction in the use of liver biopsy in > 70% of cases compared with 50% when using biomarkers (APRI, FibroTest) sequentially. Another advantage of combining two unrelated methods instead of two biomarkers is that TE provides a more direct measurement of liver structure than do biomarkers and that there is no relationship between TE and biomarkers.15
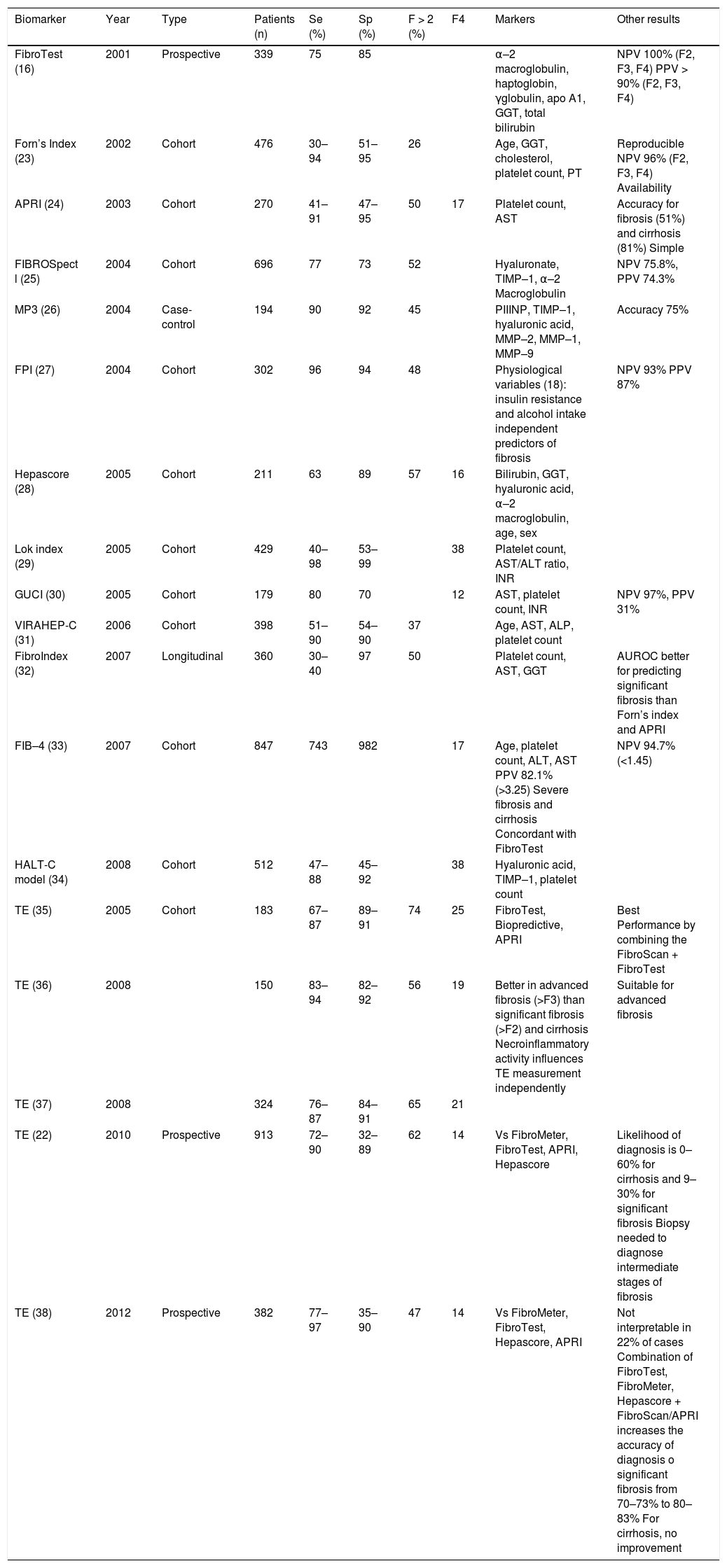
Numerous biomarkers have been proposed for use in HCV infection; the APRI and FibroTest are the methods used most widely in conjunction with TE that have been validated. The FIBROSTIC study, the largest study to date comparing TE with several patented and unpatented biomarkers and using liver biopsy as a reference, showed the equivalence of TE and serum biomarkers for the diagnosis of significant fibrosis.22 By contrast, for the diagnosis of cirrhosis, TE appears to be the most accurate method compared with currently available biomarkers and routine blood tests, and it obviates the need for liver biopsy in around 90% of cases. As a result, the combination of both methods does not seem to increase the diagnostic accuracy.15 Several studies on the diagnostic performance of these biomarkers have compared the different methods. Table 5 shows the main articles published in recent years.
- •
Recommendations: The use of noninvasive markers of HCV is recommended because they are more useful for identifying low or advanced levels of fibrosis (Class 1, Level B).
Diagnostic performance of noninvasive methods to monitor fibrosis in HCV patients.
| Biomarker | Year | Type | Patients (n) | Se (%) | Sp (%) | F > 2 (%) | F4 | Markers | Other results |
|---|---|---|---|---|---|---|---|---|---|
| FibroTest (16) | 2001 | Prospective | 339 | 75 | 85 | α–2 macroglobulin, haptoglobin, γglobulin, apo A1, GGT, total bilirubin | NPV 100% (F2, F3, F4) PPV > 90% (F2, F3, F4) | ||
| Forn’s Index (23) | 2002 | Cohort | 476 | 30–94 | 51–95 | 26 | Age, GGT, cholesterol, platelet count, PT | Reproducible NPV 96% (F2, F3, F4) Availability | |
| APRI (24) | 2003 | Cohort | 270 | 41–91 | 47–95 | 50 | 17 | Platelet count, AST | Accuracy for fibrosis (51%) and cirrhosis (81%) Simple |
| FIBROSpect l (25) | 2004 | Cohort | 696 | 77 | 73 | 52 | Hyaluronate, TIMP–1, α–2 Macroglobulin | NPV 75.8%, PPV 74.3% | |
| MP3 (26) | 2004 | Case-control | 194 | 90 | 92 | 45 | PIIINP, TIMP–1, hyaluronic acid, MMP–2, MMP–1, MMP–9 | Accuracy 75% | |
| FPI (27) | 2004 | Cohort | 302 | 96 | 94 | 48 | Physiological variables (18): insulin resistance and alcohol intake independent predictors of fibrosis | NPV 93% PPV 87% | |
| Hepascore (28) | 2005 | Cohort | 211 | 63 | 89 | 57 | 16 | Bilirubin, GGT, hyaluronic acid, α–2 macroglobulin, age, sex | |
| Lok index (29) | 2005 | Cohort | 429 | 40–98 | 53–99 | 38 | Platelet count, AST/ALT ratio, INR | ||
| GUCI (30) | 2005 | Cohort | 179 | 80 | 70 | 12 | AST, platelet count, INR | NPV 97%, PPV 31% | |
| VIRAHEP-C (31) | 2006 | Cohort | 398 | 51–90 | 54–90 | 37 | Age, AST, ALP, platelet count | ||
| FibroIndex (32) | 2007 | Longitudinal | 360 | 30–40 | 97 | 50 | Platelet count, AST, GGT | AUROC better for predicting significant fibrosis than Forn’s index and APRI | |
| FIB–4 (33) | 2007 | Cohort | 847 | 743 | 982 | 17 | Age, platelet count, ALT, AST PPV 82.1% (>3.25) Severe fibrosis and cirrhosis Concordant with FibroTest | NPV 94.7% (<1.45) | |
| HALT-C model (34) | 2008 | Cohort | 512 | 47–88 | 45–92 | 38 | Hyaluronic acid, TIMP–1, platelet count | ||
| TE (35) | 2005 | Cohort | 183 | 67–87 | 89–91 | 74 | 25 | FibroTest, Biopredictive, APRI | Best Performance by combining the FibroScan + FibroTest |
| TE (36) | 2008 | 150 | 83–94 | 82–92 | 56 | 19 | Better in advanced fibrosis (>F3) than significant fibrosis (>F2) and cirrhosis Necroinflammatory activity influences TE measurement independently | Suitable for advanced fibrosis | |
| TE (37) | 2008 | 324 | 76–87 | 84–91 | 65 | 21 | |||
| TE (22) | 2010 | Prospective | 913 | 72–90 | 32–89 | 62 | 14 | Vs FibroMeter, FibroTest, APRI, Hepascore | Likelihood of diagnosis is 0–60% for cirrhosis and 9–30% for significant fibrosis Biopsy needed to diagnose intermediate stages of fibrosis |
| TE (38) | 2012 | Prospective | 382 | 77–97 | 35–90 | 47 | 14 | Vs FibroMeter, FibroTest, Hepascore, APRI | Not interpretable in 22% of cases Combination of FibroTest, FibroMeter, Hepascore + FibroScan/APRI increases the accuracy of diagnosis o significant fibrosis from 70–73% to 80–83% For cirrhosis, no improvement |
PT: prothrombin time. GGT: gamma-glutamyl transpeptidase. Se: sensitivity. Sp: specificity. NPV: negative predictive value. PPV: positive predictive value. ALT: alanine aminotransferase. AST: aspartate aminotransferase. AP: alkaline phosphatase. TIMP: tissue inhibitor of metalloproteinase. PIIINP: procollagen type III N-terminal peptide. MMP: matrix metalloproteinases. AUROC: area under the ROC curve. APRI: aminotransferase to platelet ratio index. TE: transient elastography.
There is no specific recommendation for one method over others. Noninvasive markers of fibrosis should be selected for use according to their quality, availability, and affordability in each specific setting (Class 2b, Level C).
Treatment Of Chronic HCV GT1 in Treatment-Naïve PatientsOver the past decade, the standard of care for patients with chronic hepatitis C has comprised therapies to stimulate the immune system and to interfere nonspecifically with viral replication. The regimens used include PR given for 48 weeks to patients with HCV GT1 or for 24 weeks in those with HCV GT2 or 3. With this regimen, the SVR is 40–50% for patients with HCV GT1 and 72–84% for those with HCV GT2 or 3.8–11 A persistent SVR after long-term clearance of HCV is associated with a significant improvement in morbidity, mortality, and quality of life.39
Recently, better understanding of HCV infection, the viral life cycle, and the structure of HCV proteins and the development of a subgenomic replicon system have facilitated the development of DAAs. These new agents may substantially increase SVR rates and reduce the duration of therapy, which would greatly advance HCV therapy.40,41
It is important to mention that the clinical outcomes in patients with HCV infection are determined (among other factors) by the liver fibrosis stage. Liver fibrosis is a slow chronic continuum, for this reason there is no urgent indication to treat the HCV. Physicians should discuss with their patients the actual therapeutic options and highlight the differences between the clear indications and relative indications for treatment, as well as the risks of treatment, particularly in patients with advanced liver disease or when the treatment uses newly approved drugs, or drugs under clinical trials.
PIsThe NS3/4A serine protease of HCV plays a critical role in the HCV life cycle by cleaving most of the nonstructural proteins from the polypeptide formed during translation of the viral mRNA, and it is required for RNA replication and virion assembly. Given its essential role in the process of HCV replication, NS3/4A provides an ideal target for antiviral therapy.41
Two orally bioavailable inhibitors of the NS3/4A serine protease, BOC and TVR, have demonstrated potent inhibition of HCV GT1 replication and have substantially improved SVR rates in treatment-naïve and treatment-experienced patients. The combined use of PR plus BOC or TVR is clearly more effective than PR therapy but is more expensive and carries risks of severe AEs and drug resistance.41–43
The goal of HCV treatment is to achieve an SVR to prevent long-term complications and death.44–47 An SVR is associated with improved quality of life as measured with standard instruments such as the Short-Form–36 Health Survey. At the public health level, HCV eradication reduces transmission risk.
In patients without advanced fibrosis before treatment, an SVR represents a cure. For those with cirrhosis, an SVR is a virological cure that is associated clearly with improved outcomes, although liver cancer may still develop.44–47 Chronic hepatitis C patients infected with HCV GT1 are considered difficult to treat and are therefore the primary focus for the development of DAAs.
BOCIn registration trials, the combined use of BOC with PR significantly improved SVR rates in HCV GT1 treatment-naïve patients, relapsers, and partial responders. Based on these results, BOC has been approved to treat these groups as well as nonresponder patients. All phase III trials with BOC were designed with a lead-in period with dual therapy with PR before the administration of BOC, based on results obtained in phase II studies. The rationale for this strategy is based on the fact that both PR reach steady-state concentrations by week 4, and therefore, using this approach, the PI is added when high drug levels have been reached and the immune system has been stimulated. This strategy serves to lower HCV RNA levels before exposure to the PI, thereby reducing the risk of viral breakthrough or resistance to BOC.
The phase III SPRINT-2 trial, which included 1,097 patients, was designed to evaluate the impact of triple therapy with PR plus BOC in two cohorts of patients: 940 Caucasian and 159 black HCV GT1 treatment-naïve patients.8 Most of the patients were infected with HCV GT1a, with nearly 75% of blacks infected with this subtype. Most patients had a high HCV RNA level (> 400,000 IU/mL) and 7–11% had METAVIR F3 or F4 disease. All patients received a 4-week lead-in phase with PR and weight-based RBV (Ribavirin). After 4 weeks, patients were randomized to one of three treatment groups. Patients assigned to group 1 (control group) received PR for 44 weeks after the lead-in period, beginning at week 5. Patients assigned to group 2 (RGT group) received PR, and BOC for a total of 24 weeks after the lead-in period. If HCV RNA levels were undetectable (< 10–15 IU/mL) from week 8 through week 24, therapy was considered complete and all treatment was stopped at week 28. If HCV RNA levels were detectable at any visit from week 8 up to but not including week 24 (slow virological response), PR (and placebo) was continued through week 48. Patients assigned to group 3 (fixed-duration therapy) received PR and BOC for a total of 44 weeks after the lead-in dual-treatment period. The BOC dose was 800 mg, given by mouth three times per day with food; PR was administered subcutaneously at a dose of 1.5 μg/kg body weight once weekly; and weight-based oral RBV was administered at a total dose of 600–1,400 mg/day in divided doses, in the morning and evening. All patients were followed through week 72. In all three groups, the study treatment was discontinued for all patients with a detectable HCV RNA level at week 24, according to a standard stopping rule.
SVR rates were significantly higher in the BOC groups (Caucasian and black patients): 63% in the fixed-duration therapy group, 66% in the RGT group, and 38% in the control group. The SVR rates in Caucasians were 68% in the fixed-duration group, 67% in the RGT group, and 40% in the control group. The SVR rates were lower in black patients: 53% in the fixed-duration group, 42% in the RGT group, and 23% in the control group. Fifty-four percent of Caucasian patients who received BOC experienced an RVR, with HCV RNA undetectable at week 8. This interval was selected because of the 4-week lead-in period. By contrast, only 20% of black recipients of BOC experienced an RVR. BOC-treated patients who achieved an RVR had a high probability of achieving an SVR; the SVR rates were 89–91% for non-black patients and 78–82% for black patients. The SVR rates were lower in slow responders (HCV RNA detectable at week 8 but undetectable at week 24) than in those with an RVR. The SVR rates also appeared to be slightly better in the BOC fixed-duration group than in the BOC RGT group: 43 vs. 37%, respectively, for nonblack patients and 32 vs. 28%, respectively, for black patients. The lower response rate found for slow responders prompted the FDA (food drug administration)-approved label to include extending BOC therapy to week 36 and then continuing with PR in this group. Patients with F3 or F4 stage fibrosis had higher SVR rates if they received the fixed 48-week course of BOC with PR compared with the RGT group: 52 vs. 41%, respectively.48
This trial showed that the SVR rates were higher for all BOC-containing regimens across all the pretreatment variables that had been identified in previous studies to influence the response to dual therapy, including high pretreatment HCV viral load level, advanced fibrosis, and race. The rates of SVR in the BOC groups, for both Caucasian and black patients, were similar between the RGT and fixed-duration groups and were significantly higher when RGT was compared with dual PR treatment.
TVRIn the registration trials, combined treatment using TVR with PR significantly improved the SVR rates in HCV GT1 treatment-naïve patients, relapsers, partial responders, and nonresponders. Based on these results, TVR has been approved to treat these patients.
Two phase III trials (ADVANCE and ILLUMINATE) were designed to evaluate the impact of triple therapy with PR plus TVR in treatment-naïve patients with HCV GT1.49,50 The ADVANCE trial, which included 1,088 patients, was a randomized study designed to evaluate safety and efficacy, and two other aspects. The first was to explore whether 8 weeks rather than 12 weeks of TVR could reduce AEs, specifically rash, by preserving the virological response. The second was to investigate the SVR in those patients with an RVR when treatment was stopped at 24 weeks. Patients were assigned to one of three groups:
- •
TVR plus PR for 8 weeks followed by PR for 24 or 48 weeks (group T8PR).
- •
TVR plus PR for 12 weeks followed by PR for 24 or 48 weeks (group T12PR).
- •
PR for a total duration of 48 weeks (group PR).
The TVR dose was 750 mg, given by mouth three times per day with food (a meal containing 20 g of fat); PR was given at a dose of 180 μg, injected subcutaneously once weekly; and RBV was given at a dose of 1,000 mg per day for patients weighing < 75 kg or 1,200 mg per day for patients weighing ≥ 75 kg. In contrast to the BOC regimens, all three drugs were started on day 1 of therapy.49 Patients who achieved an extended RVR (eRVR) (undetectable HCV RNA at both weeks 4 and 12) stopped treatment at 24 weeks. Those patients who failed to attain an eRVR were treated for 48 weeks.
The SVR rates were 75% for the T12PR group, 69% for the T8PR group, and 44% for the PR group. An RVR was observed in 68% of T12PR patients, 67% of T8PR patients, and 9% of PR patients. An eRVR was achieved in 58%, 57%, and 8% of patients, respectively; both the T12PR and T8PR groups were able to stop therapy at week 24. The SVR rates were higher for patients who achieved an eRVR: 89%, 83%, and 97% in the T12PR, T8PR, and PR groups, respectively. Patients who did not achieve an eRVR had SVR rates of 54%, 50%, and 39% in the T12PR, T8PR, and PR groups, respectively. Similar to the data from the study using BOC, patients with F0-F2 fibrosis had a higher SVR rate than did patients with F3 or F4 fibrosis (78 vs. 62% in the T12PR group).39
In a retrospective analysis of the ADVANCE trial, a substudy of around 42% of the study population, all of whom were white, showed that the addition of TVR to PR increased SVR rates across all IL28B genotypes. Patients with the IL28B CC genotype achieved higher SVR rates compared with patients with the CT or TT genotype. The SVR rates in patients with the CC genotype were 90% with 12 weeks of TVR-based therapy, 84% with 8 weeks of TVR-based therapy, and 64% with PR alone. Among patients with the CT genotype, the SVR rates were 71%.51 The ADVANCE study strongly suggested that 24 weeks of total therapy is sufficient in patients with an eRVR, but that in those with a slow virological response or cirrhosis, a total of 48 weeks of therapy is necessary.
The ILLUMINATE study was designed to confirm that shortened treatment duration, based on RGT, was not inferior to fixed-treatment duration for HCV GT1 treatment-naïve patients with chronic hepatitis who achieved an eRVR. Those who achieved an eRVR were randomized at week 20 to receive either 24 or 48 weeks of PR therapy. The SVR rates among patients who achieved an eRVR (65%) were 92% for 24 weeks vs. 88% for 48 weeks of therapy. These findings confirmed the noninferiority of the 24-week RGT therapy.50
The use of BOC or TVR in association with PR can be considered the new standard of care for HCV GT1-infected patients. In the era of PI therapy, an early response remains a critical determinant of treatment outcome.
The FDA has recently added a boxed warning to the drug label saying that TVR combination treatment should be stopped immediately in patients who develop a rash and systemic symptoms, or a progressive rash, and that these patients should receive treatment. Systemic symptoms associated with these serious skin reactions include fever, nausea, diarrhea, mouth sores or ulcers, facial swelling, red eyes, and liver swelling.52
- •
Recommendations:
- °
All patients with chronic HCV infection should be evaluated for HCV antiviral treatment (Class 2, Level B).
- °
All treatment-naïve patients with compensated disease because of HCV should be considered for therapy (Class 1, Level A).
- °
The combination of BOC or TVR with PR (triple therapy) is the new standard of care for HCV GT1-infected patients (Class 1, Level A).
- °
The protease inhibitors BOC and TVR should not be used without PEG-IFN (pegylated interferon) and a weight-based dose of RBV (Class 1, Level A).
- °
The recommended dose of BOC is 800 mg administered with food three times per day (every 7–9 h) together with PEG-IFN and a weight-based dose of RBV, for 24–44 weeks; this should always be preceded by a 4-week lead-in period with PR alone (Class 1, Level A).
- °
Patients without cirrhosis treated with BOC and PR (preceded by 4 weeks of a lead-in period) with undetectable HCV RNA at weeks 8 and 24 may be considered for a shortened duration of treatment of 28 weeks in total (lead-in period of 4 weeks with PR, followed by 24 weeks of triple therapy) (Class 2a, Level B).
- °
Patients with cirrhosis treated with BOC and PR should receive therapy for 48 weeks (Class 2b, Level B).
- °
The stopping rules for BOC are as follows: treatment with BOC and PR should be stopped if the HCV RNA level is > 100 IU/mL at week 12 of treatment or is detectable at week 24 or at any time thereafter, and if HCV RNA rebounds at any time (≥ 1 log10 increase from the nadir HCV RNA level) (Class 2a, Level B).
- °
The recommended dose of TVR is 750 mg administered with food (a meal including 20 g of fat) three times per day (every 7–9 h) together with PEG-IFN and weight-based RBV, for 12 weeks; this should be followed by an additional 12–36 weeks of PR (Class 1, Level A).
- °
Patients without cirrhosis treated with TVR and PR with undetectable HCV RNA at weeks 4 and 12 should be considered for a shortened duration of therapy of 24 weeks (Class 2a, Level A).
- °
Patients with cirrhosis treated with TVR and PR should receive therapy for 48 weeks (Class 2b, Level B).
- °
Stopping rules for TVR: treatment with TVR and PR should be stopped if the HCV RNA level is > 1,000 IU/mL at weeks 4 or 12 of treatment and/or is detectable at week 24 or at any time thereafter, and if the HCV RNA level rebounds at any time (≥ 1 log10 increase from the nadir HCV RNA level) (Class 2a, Level B).
- °
If BOC or TVR is suspended, it should not be restarted.
- °
If virological failure occurs with a BOC- or TVR-containing regimen, the PI involved must not be substituted by another PI (Class 1, Level C).
- °
Those patients under treatment with TVR who develop a rash and systemic symptoms, or a progressive rash, should stop treatment (Class 3, Level C).
- °
Current information shows the superiority of triple therapy. There is no scientifically based information to indicate the need to delay new therapies or to use PR. The patient should be informed of the potential benefits, risks, and costs in each specific setting (Class 2a, Level C).
- °
About half of patients infected chronically with HCV fail to achieve an SVR when treated with PR.53 This situation is particularly problematic for patients with advanced liver disease because no maintenance therapy has been proven to halt disease progression in patients who do not achieve an SVR.7 Fortunately, HCV treatment is progressing rapidly, with the first-generation PIs BOC and TVR already available in many parts of the world for use in combination with PR for both treatment-naïve and treatment-experienced patients with HCV GT1.8,9,49,54
Considering that better regimens will likely be available in the next few years, one of the major challenges in the field at present is how to select which HCV GT1-infected patients whose previous therapy with PR failed should be retreated, and which should wait for second-generation DAAs. It seems that, among many important variables, disease stage and type of treatment failure can be useful when making these decisions.7,54 In selected cases, the type of response to a 4-week lead-in therapy of PR could help identify candidates for retreatment.54,55
Types of Treatment FailureThere are four types of treatment failure:44
- •
Null response.
- •
Partial response.
- •
Breakthrough.
- •
Relapse.
The nomenclature and with failure to achieve an SVR after HCV definitions currently applied for the different types of outcome associated treatment with PR are described in table 6 and figure 1.
Types of HCV treatment failure with PEG-IFN and RBV.
| Nomenclature | Definition |
|---|---|
| Null responders | Patients with a reduction of <2 log10 in HCV RNA level after 12 weeks of therapy. |
| Partial responders | Patients with a reduction of ≥2 log10 but with detectable serum HCV RNA during therapy. |
| Breakthrough | Patients with a return of HCV RNA during treatment after having a previously undetectable HCV RNA level, usually occurring because of dose reduction or missing periods of PEG-IFN and/or RBV administration related to poor treatment tolerability, not a true lack of interferon sensitivity |
| Relapsers | Patients with an undetectable serum HCV RNA level during treatment and at the end of treatment, but with a rebound in HCV RNA level after the end of treatment. |

Among patients who previously failed to respond to a PR regimen, the probability of achieving an SVR after retreatment with TVR55 or BOC7 is usually related to two main variables:
- •
Type of treatment failure, and
- •
Fibrosis stage.
Interestingly, contrary to what has been shown for treatment-naïve patients, there is no compelling evidence that IL28B polymorphism can effectively predict an SVR among patients who previously failed PR treatment. Although IL28B polymorphism is a good surrogate marker for IFN sensitivity, among patients who have already been exposed to an IFN-based regimen, the type of response is a much better measure of IFN sensitivity than is IL28B genotype testing.7,54 For this reason, relapsers are the best candidates for retreatment because these patients were able to clear HCV RNA from the circulation during the first treatment, albeit transiently, which is a clear sign of IFN sensitivity.7,54 IL28B genotype testing could still be used in treatment-experienced patients for whom the previous treatment history is either incomplete or unavailable.
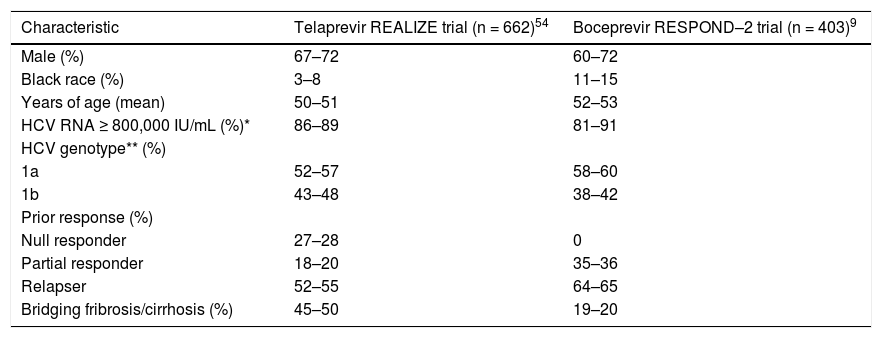
Probability of an Svr Upon Retreatment of HCV GT1-Infected PatientsRetreatment of HCV GT1-infected patients whose previous therapy failed was addressed in two phase III trials, one with TVR (REALIZE study)54 and the other with BOC (RESPOND–2 study).9 Patients with breakthrough were excluded from these trials probably because this situation is more often related to drug tolerability issues that lead to irregular dosing. The probability of achieving an SVR in the REALIZE and SPRINT-2 trials was analyzed according to the patients’ prior response to treatment (Figures 2 and 3, respectively) and disease stage (Table 7 and Figure 4, respectively). The SVR rate was usually 5–10% lower in patients infected with HCV GT1a compared with those infected with GT1b for all categories of previous treatment failure, but SVR was more pronounced in those patients who previously had a partial or null response.7,54


Design of the boceprevir RESPOND–2 study. Patients with detectable HCV RNA at week 12 were considered treatment failures and discontinued treatment. RGT: response-guided therapy. Peg-IFN alfa–2b: 1.5 μg/kg/week. RBV: 600–1,400 mg/day. BOC: boceprevir 800 mg every 7–9 h. Bacon, et al. 2011.9 Boceprevir EU SmPc.
Key baseline characteristics in the REALIZE and RESPOND–2 trials.
| Characteristic | Telaprevir REALIZE trial (n = 662)54 | Boceprevir RESPOND–2 trial (n = 403)9 |
|---|---|---|
| Male (%) | 67–72 | 60–72 |
| Black race (%) | 3–8 | 11–15 |
| Years of age (mean) | 50–51 | 52–53 |
| HCV RNA ≥ 800,000 IU/mL (%)* | 86–89 | 81–91 |
| HCV genotype** (%) | ||
| 1a | 52–57 | 58–60 |
| 1b | 43–48 | 38–42 |
| Prior response (%) | ||
| Null responder | 27–28 | 0 |
| Partial responder | 18–20 | 35–36 |
| Relapser | 52–55 | 64–65 |
| Bridging fribrosis/cirrhosis (%) | 45–50 | 19–20 |

SVR rates with telaprevir in the REALIZE trial according to prior response type. *p < 0.001 vs. PR48. SVR, considered virologc cure, was defined as HCV RNA < 25 IU/mL at last observation within he week 72 visit window. In case of missing data, the last HCV RNA data point from week 12 of follow-up onwards was used. Telaprevir EU SmPc.
Retreatment with PR is the only option for patients infected with HCV GT2 or 3 whose previous therapy failed. Relapsers have a > 50% chance of an SVR when retreated for longer period of time.56 However, partial or null responders have almost no chance of an SVR and should probably wait for future therapies. In a few selected cases, however, a possible reason for treatment failure could be identified and corrected, increasing the chance for an SVR upon retreatment.
Treatment-Naïve Cirrhotic PatientsThe main objectives of treatment in cirrhotic patients are to prevent disease complications, allow regression of fibrosis, and avoid reinfection of the graft in those patients who will receive a liver transplant.57
When the SVR rate has been evaluated in cirrhotic patients, especially in those with decompensated disease, it has been found to be lower than in patients with chronic hepatitis without cirrhosis.8,49,58Cirrhotic patients also have a higher probability of experiencing treatment complications. In Child-Pugh A cirrhotic patients, 30–40% will achieve an SVR; and patients who do not will have a higher probability of developing hepatocellular carcinoma (HCC) and a higher mortality rate.58–59 The SVR rate is higher (> 50%) in patients with cirrhosis and HCV GT1 treated with either BOC or TVR in combination with PR compared with PR alone.8,49 These patients are not candidates for RGT,8,49,50 and it has been suggested that they need to receive therapy for 48 weeks, although this is based on limited data and requires further study.60,61
Regarding treatment benefits, an evaluation of studies of patients with cirrhosis showed that SVR rates are lower in patients with cirrhosis than in those without and that treatment success rates are lower for decompensated cirrhotic patients because of the poor response and increased probability of developing complications.62,63 There is a paucity of information regarding treatment with DAAs of patients with decompensated cirrhosis. Treatment with triple combination therapy is clearly indicated in patients with compensated cirrhosis and HCV GT1, with the conditions that their starting blood cell count must be sufficient for them to tolerate the decrease in cell count induced by the treatment and that there are no other contraindications. Drug-drug interactions require careful consideration. The therapy may be contraindicated in advanced cirrhotic patients with Child-Pugh B or C classification because of the possibility of decompensation and death.60
- •
Recommendations:
- °
Patients with compensated Child-Pugh A cirrhosis must be referred for evaluation and must receive the usual treatment (Class 1, Level A).
- °
Patients with compensated Child-Pugh A cirrhosis and HCV GT1 must receive triple combination therapy (Class 1, Level A), preferably for 48 weeks (Class 2, Level C).
- °
Patients with Child-Pugh B cirrhosis awaiting transplantation could be treated if the virus is GT2; if treated, patients with Child-Pugh B or C liver disease after compensation should be followed carefully in experienced centers (Class 2, Level B).
- °
Patients with decompensated Child-Pugh B or C liver disease should be referred to a transplantation unit for evaluation (Class 1, Level A).
- °
Liver transplantation offers an effective treatment that significantly reduces morbidity and mortality among patients with HCV. However, hepatic graft reinfection is universal in posttransplantation HCV-infected patients because of the advanced liver disease related to HCV. Reinfection is almost inevitable in patients with measurable viral RNA given a liver transplant. The natural history of hepatitis C is more aggressive after liver transplantation than in immune-competent patients, and other adverse risk factors may be present (e.g., metabolic syndrome).64 Graft cirrhosis has been reported in almost 30% of patients 5 years after liver transplantation, and their survival rate is significantly lower than that of HCV-negative patients.59,65–68
Antiviral therapeutic strategies to achieve an SVR could be given before or after transplantation. The aims of antiviral therapy before transplantation are to achieve an SVR and clearance of viral RNA at the time of transplantation, to prevent recurrence, and to stop disease progression.69–71
All patients on the transplant list with Child-Pugh A classification, mainly those with HCC, are recommended to receive antiviral therapy. This treatment is contraindicated in those with Child-Pugh C classification or a score of > 18 in the Model for End-Stage Liver Disease. The success in clearing HCV is about 15–20% in patients infected with HCV GT1 and 20–35% in those with GT2 or 3.63,70–72
The post-transplantation antiviral treatment is used to improve the survival of the patient and the graft when an SVR is achieved. The therapeutic strategies include preventive treatment or treatment when HCV is established. A preventive strategy seems attractive because treatment is commenced while the viral level remains low and before the graft is damaged, and thus higher rates of SVR would be expected in patients treated this way. Nevertheless, in practice, only 40–60% of patients are candidates to receive treatment because these patients receive high-dose immunosuppression with consequent pancytopenia, mild renal dysfunction, and the presence of additional medical conditions during the early posttransplantation phase.39,66,67,73–75
The efficacy of treatment for established recurrent HCV depends on the treatment type and duration and the ideal time to start treatment (acute or chronic phase). Prophylactic antiviral therapy has no clear benefits in terms of HCV recurrence and patient or graft survival. In one study, an SVR was achieved in 22.2% of patients given prophylactic antiviral therapy.76 In daily practice, treatment is generally started in the chronic phase, especially when progression of fibrosis is observed.77–80 Monotherapy with IFN or PEG-IFN is not recommended because it is associated with a low SVR rate. The standard therapy, independent of genotype, comprises PR for 48–52 weeks. This provides higher response rates (SVR rates of 20–30% for HCV GT1 and 40–50% for HCV GT2 or 3) but is not well tolerated by patients in the early posttransplantation phases, and the dose needs to be reduced in most patients. The main causes of dose reduction and premature discontinuation of treatment are AEs such as blood disorders, neuropsychiatric disorders, infections, thyroid abnormalities, and poor clinical tolerance. The development of cellular rejection (6%) or chronic rejection (1%) can require discontinuation of treatment. Most patients require erythropoietin, granulocyte colony-stimulating factor, and blood transfusions.77–80 The predictive factors for treatment response are infection with HCV non-GT1, low initial viral load, and mild fibrosis. Polymorphisms in the IL28B genotype can affect the SVR. The genetic profiles of both the recipient and the donor play a role. The RVR (at 4 weeks) and eVR (at 12 weeks) are the best SVR predictors.81–82
BOC and TVR are both substrates and inhibitors of the hepatic enzyme cytochrome P450 3A4, the enzyme responsible for the metabolism of the calcineurin inhibitors (CNIs) cyclosporin and tacrolimus, and the drug transporter P-glycoprotein. This relationship predisposes these agents to many drug interactions and will place the patient at potential risk for both CNI toxicity (when a PI is given with a CNI) and acute rejection (when a PI is stopped). For transplant recipients, very close monitoring and adjustment of CNI levels is critical during PI-based triple therapy.83–85 CNIs are only just beginning to be tested and at present should not be used as regular therapy in liver transplant recipients, with the possible exception of patients participating in a clinical trial.85–86 The AEs can be serious and may include increased risk of infection, rejection, dermatological changes, or death.87 The dose of PR is decreased or stopped early in most post-transplantation patients. Most require erythropoietin, granulocyte colonystimulating factor, and blood transfusion. The drugdrug interactions (cyclosporin A or tacrolimus with BOC or TVR) can be managed by close monitoring.87
Triple therapy (PEG-INF, RBV, and TVR or BOC) increases the probability of an SVR in about 70% of treatment-naïve persons and relapsers after PEG-INF/RBV and represents an important advance in the treatment of chronic HCV.84,87 The prospects for triple therapy seem to be optimistic, with the possibility of increasing the number of viral-negative patients, and knowledge about the medications allows a better understanding of their effects and possible patient adverse reactions.
Multicenter, prospective studies with more patients and longer follow-up are needed to reach firm conclusions before triple therapy can be recommended.
- •
Recommendations:
- °
Treatment of HCV in patients who receive a liver transplantation is indicated only in patients with adequate supervision of therapy administration and of AEs (Class 1, Level A).
- °
The therapeutic regime of choice in patients should be PEG-INF/RBV (Class 1, Level A).
- °
The duration of antiviral therapy recommended in post-transplantation patients is 48–52 weeks independently of the HCV genotype (Class 1, Level A).
- °
Post-transplantation patients should receive erythropoietin and granulocyte growth factors so that the RBV dosage does not need to be reduced (Class 1, Level B).
- °
All patients on the transplant list with Child-Pugh A classification, mainly those with HCC, are recommended to receive antiviral therapy. The treatment is contraindicated in those with Child-Pugh C classification or a Model for End-Stage Liver Disease score > 18 (Class 1, Level B).
- °
A preventive strategy using antiviral therapy is not recommended (Class 1, Level B).
- °
Prophylactic antiviral therapy is not recommended (Class 1, Level A).
- °
The predictive factors for a treatment response are HCV non-GT1, a low initial viral load, and the presence of mild fibrosis (Class 1, Level A).
- °
Polymorphisms in the IL28B gene affect the SVR. The genetic profiles of both the recipient and the donor play a role in the antiviral response (Class 1, Level B).
- °
The RVR (at 4 weeks) and eVR (at 12 weeks) are the best predictors of an SVR (Class 1, Level A).
- °
Triple therapy (PEG-INF/RBV and TVR or BOC) increases the SVR rate. This therapy is beginning to be tested and, at present, should not be used as a regular therapy in liver transplant recipients, with the possible exception of those enrolled in a clinical trial (Class 2a, Level C).
- °
HCV is a positive-strand RNA virus with a genome of about 10,000 nucleotides. The virus displays significant genetic heterogeneity, with six main genotypes (designated 1–6) and > 50 subtypes (denoted by letters) that differ mainly in their amino acid sequences.88
HCV Quasispecies and Resistant VariantsHCV has two steps at which mutations can be introduced: during the production of the negativestrand RNA intermediate and during the synthesis of the new positive-strand RNA genome from the negative-strand template. The key protein responsible for viral RNA synthesis is HCV NS5B (the catalytic subunit of the replication complex), which has RNA- /dependent RNA polymerase activity.89–90 HCV replicates at a rapid rate: it is estimated to produce and clear about 1012 virion particles per day, with a mean half-life of free virions of 2–3 h.91 This situation may generate single, double, and possibly triple mutants (i.e., viruses with one, two, or three amino acid substitutions, respectively) every day, which may alter the drug target and decrease the virus’s susceptibility to the inhibitory activities of drugs. These drug-resistant variants preexist as minor populations in many treatment-naïve infected patients and are detected rarely by low-sensitivity techniques before therapy because the amino acid substitution(s) also reduce the replicative capacity in the absence of a given antiviral drug (i.e., they have a reduced viral replication fitness).92–93
Monotherapy studies with DAAs with a low barrier to resistance (small number of mutations required by a virus to become drug resistant) including NS3/4A protease inhibitors, the non-nucleoside inhibitors of HCV RNA-dependent RNA polymerase, and NS5A inhibitors have shown early virological breakthrough because of the selection and outgrowth of fit, drug-resistant viral populations.94 This high variability represents a challenge for therapeutic antiviral strategies because the virus may rapidly evade the host immune responses and the effects of antiviral drugs.
Antiviral Treatments for HCV and Viral ResistanceBefore the introduction of DAAs, the standard of care for chronic HCV infection was PR. No selection of resistant viral isolates followed by a viral breakthrough has conclusively been demonstrated for these drugs.95 In view of the lack of effectiveness of the current therapy, many molecules have been tested in the search for new anti-HCV therapies. Several new selective inhibitors of HCV proteins, the DAAs, have been in development, including HCV NS3-4A protease, NS5B polymerase, and NS5A inhibitors.88,91,96 Although these agents have demonstrated potent antiviral effects, monotherapy is complicated by rapid virological breakthrough because of the selection of drug-resistant mutants.62,93,97 Since 2011, the new standard of care for HCV GT1-infected patients is the triple combination therapy of PR and an HCV NS3/4A PI (BOC or TVR).7 An important limitation of these DAAs is their low genetic barrier for resistance, resulting in drug-escape mutants during long-term treatment because of their general mechanism of action.98
A recent analysis of the long-term persistence of resistant mutations within the HCV NS3 protease seen at the end of antiviral treatment in phase Ib studies (monotherapy) has shown that, after a mean follow-up of 4.2 years, 2 of 14 patients treated with TVR and 4 of 14 patients treated with BOC retained variant mutations.99 Further studies to identify potential persistence of resistant variants using more sensitive techniques, such as ultra-deep sequencing methods, are needed to understand this problem better.
Resistance is determined by several factors, including:
- •
Drug-selective pressure (potency and concentration at the site of replication).
- •
The genetic barrier to resistance (number of mutations required for complete loss of activity); and
- •
The rate of replication of resistant strains (viral fitness).100
The detection of viral resistance depends on the sensitivity of the assay. Current methods to characterize viral resistance include genotypic and phenotypic assays. At present, there are no commercial assays to measure resistance that are available for routine clinical practice, but ‘home brew’ molecular methods have been developed in reference laboratories.
Sensitive methods include ultra-deep sequencing methods, such as pyrosequencing, which can detect minor resistant populations down to < 1%, and the TaqMan® mismatch amplification mutation assay, which is highly reproducible and linear over a wide range of mutant levels (0.01–100%) and can consistently detect variants al the level of 0.1%.94 However, these tools are costly and time consuming.
Cross-resistance occurs when resistant mutations are selected using more than one drug with a common antiviral binding position. There is no cross-resistance between classes of compounds that target different mechanisms of action.8 Thus, future drug regimens for treatment of HCV infection are likely to include combinations of multiple classes of inhibitors (e.g., NS3/4A protease, NS5B polymerase nucleoside, NS5B polymerase non-nucleoside, and NS5A inhibitors) precisely to avoid resistant mutations.
Preexistent Viral Variants and the Clinical Impact of ResistanceThe presence of variants resistant to DAA that inhibit NS3/4A (TVR-resistant variants) or NS5A in baseline samples of treatment-naïve patients receiving a TVR-based regimen did not affect the SVR.101
Antiviral response pattern to DAA therapy and resistanceTo minimize the implications of developing resistance, treatment with a DAA should be stopped upon detection of viral breakthrough to prevent the further evolution of resistant variants (a stopping rule).
Monitoring HCV RNA During TreatmentA sensitive assay with a broad quantification range (e.g., real-time PCR assay) should ideally be used.102,103 The lower limit of quantification of an HCV RNA assay is the lowest HCV RNA concentration that is within the linear range of the assay; i.e., the smallest amount of HCV RNA that can be detected and quantified accurately. The lower limit of detection is the lowest actual amount of HCV RNA that can be detected with 95% probability to determine the presence or absence of the virus.
Quantitative assays are required to make treatment decisions.102,103 Quantitative HCV RNA assays with a lower limit of quantification near 25 IU/mL and a lower limit of detection of 9.3–15 IU/mL (such as the COBAS TaqMan 2.0) should be used repeatedly when managing patients who receive TVR- or BOC-based triple therapy.
- •
Recommendations:
- °
Given that HCV variants resistant to DAAs are likely to preexist in treatment-naïve HCV patients and that their presence is not necessarily associated with treatment failure, there is currently a limited role for the evaluation of these variants before beginning antiviral treatment (Class 3, Level C).
- °
Treatment-naïve patients with viral populations containing NS3/4A-resistant variants achieve the same SVR rate as do patients with no resistant variants before treatment, in the setting of adequate PEG-IFN responsiveness (Class 1, Level B).
- °
Patients on PI-based therapy should undergo close monitoring of HCV RNA levels using a very sensitive assay, and the PI should be discontinued immediately if virological breakthrough occurs (> 1 log10 increase in serum HCV RNA level from the nadir) because the dominant virus at this time is most often resistant to the PI (Class 1, Level A).
- °
During the follow-up after discontinuation of DAAs caused by virological breakthrough, in most cases, there is a reversion to wild-type HCV. In the absence of alternative antiviral treatments, resistance testing is not recommended (Class 3, Level C).
- °
Patients who fail to have a virological response, experience virological breakthrough, or relapse after treatment with one PI should not be retreated with another licensed PI because of the potential for cross-resistance (Class 2a, Level C).
- °
Treatment with triple therapy can lead to significant AEs that necessitate dose reduction or discontinuation of treatment. Early recognition and intervention can help clinicians ensure that patients are able to complete therapy where possible and achieve the goal of viral eradication. Although every patient will experience AEs differently, close monitoring through frequent visits and laboratory testing and a systematic approach to the patient’s management can be very helpful.
AnemiaIn clinical trials, a significantly higher proportion of patients developed anemia with PI-containing regimens compared with dual therapy. However, anemia develops more frequently with BOC than with TVR. In the SPRINT–2 trial, a hemoglobin level < 10.0 g/dL was reported in 45 and 41% of patients in the two groups using BOC, whereas it was reported in only 26% in the PR group.8 However, a hemoglobin level < 8.5 g/dL was observed in 5 and 9% in the two BOC groups vs. 4% in the control group. Erythropoietin-alpha (EPO) was used in 43% of the patients, and 3% of them required a blood transfusion.8,9,54
In the ADVANCE trial, a hemoglobin level < 10 g/dL was observed in 36% of patients in the TVR groups compared with 14% in the PR group. A hemoglobin level < 8.5 g/dL was observed in 9% of the TVR-treated patients compared with 2% in the PR-treated patients. Anemia had led to premature discontinuation in 1–3% of TVR-treated patients.49 In the REALIZE study, anemia occurred in 30–36% of patients in the TVR-treated groups compared with 15% in the control group.54 In studies of TVR, anemia was managed only by RBV dose reduction because EPO was not allowed. TVR treatment was discontinued in 2–4% of patients, and the entire treatment had to be stopped in about 1% of patients.49,54 Blood transfusion was necessary in 12% of TVR-treated patients and in 5% of patients in the control group. In these phase III studies, the maximum decrease in hemoglobin level occurred at around the second month of treatment.49,54 It is important to mention that studies performed in actual patients included a higher proportion of those with cirrhosis or older patients and showed higher rates of anemia, blood transfusion, and EPO use than in patients included in randomized clinical trials.104
Anemia managementAnemia can be managed with RBV dose reduction as the first action. EPO can be administered and even blood transfusion may be required. Eventually, anemia resolves with discontinuation of the PI. The dose of the PI cannot be reduced under any circumstance because this increases the risk of resistance.
Among BOC-treated patients, SVR rates are identical to those given a reduced RBV dose and those given EPO.105 RBV dose reduction less frequently requires a second intervention to control anemia.105 Despite early information about reduced treatment efficacy,106,107 with BOC therapy, SVR rates are similar regardless of the timing of the first RBV dose reduction, number of RBV dose reduction steps, and lowest RBV dose received for anemia management.108 In TVR-treated patients, RBV dose reduction also has no effect on SVR rates.109 The development of anemia is associated with higher SVR rates in patients treated with PI-based therapy.110
There are no clear recommendations for EPO use in HCV treatment-induced anemia, and this indication has not been approved in many countries. The French Drug Administration has issued specific guidelines for EPO treatment in HCV treatment-induced anemia.111
Collectively, these data suggest that RBV dose reduction is the most appropriate first-line strategy for management of anemia. It may be necessary to maintain the full dose of RBV until HCV RNA becomes undetectable. If anemia occurs when HCV RNA is undetectable, the RBV dose may be reduced by increments of 200 mg daily. EPO can be used when RBV dose reduction is not enough to control decreasing hemoglobin levels. Blood transfusion may be required and can be effective, particularly for patients with cirrhosis. If RBV must be discontinued, the PI must also be discontinued because there is a high risk of the development of drug-resistant variants when a DAA is used with PEG-IFN only.106,112 Once BOC or TVR has been stopped, it should not be restarted. BOC and TVR dosing must not be reduced.
- •
Recommendations:
- °
Early and close monitoring of the hemoglobin level is needed, especially during PI treatment and when the hemoglobin level is < 10 g/dL (Class 1, Level C).
- °
Management of anemia may include RBV dose reduction (Class 1, Level A), EPO use ( Class 2a, Level C), and/or red blood cell transfusion (Class 1, Level C). The PI dose should never be reduced, and once suspended should never be reinitiated (Class 1, Level A).
- °
Dermatological AEs are commonly seen with TVR use. Reported rates for TVR vs. PR were 56 vs. 34%, respectively, for rash and 47 vs. 28%, respectively, for pruritus.113 In phase III trials, 5–7% of patients discontinued TVR because of rash.49,54 The symptoms usually resolve following treatment discontinuation, but it could take weeks for complete resolution. Rash lesions are eczematous squamous eruptions with pruritus. Rash is seen in 50% of patients within the first 4 weeks of therapy; the symptoms are predominantly mild and are generally nonprogressive.113 However, the possibility of rash extension around the lesions should be checked.
Rash is classified by severity into three grades:
- •
Grade 1. Or mild and localized, involving < 25% of body surface area.
- •
Grade 2. Or moderate and diffuse, involving up to 50% of body surface area.
- •
Grade 3. Or severe and generalized, involving either > 50% of the body surface area and accompanied by systemic signs or symptoms or by severe cutaneous adverse reactions.
In these patients, rash episodes were graded 1, 2, and 3 in 37%, 14%, and 5% of patients, respectively. Extension of the skin reaction is unusual; progression to a more severe grade of rash occurred in only in 8% of cases, and severe cutaneous adverse reaction occurred rarely.42
In BOC-treated patients, the rates of dermatological AEs were similar to those in the control group: 17% in treatment-naïve and 16% in treatment-experienced patients vs. 19% and 6%, respectively, in PEG-IFN-treated controls.10 However, a recent report from the FDA alerts clinicians to the possibility of serious skin reactions in patients treated with BOC.
Rash management planThe rate of discontinuation of all study drugs as a result of dermatological AEs was lower in TVR phase III trials than in phase II trials43 following the incorporation of a rash management plan into the study protocols.49,54 Grade 1 or 2 (mild or moderate) rash does not require treatment discontinuation and can be treated primarily using emollients/moisturizers and topical corticosteroids. Topical or systemic antihistaminic drugs (including diphenhydramine, hydroxyzine, levocetirizine, or desloratadine) can be used. Patients should be advised to wear loose-fitting clothes and to limit their exposure to sun and heat. Grade 3 rash requires immediate discontinuation of TVR. The symptomatic treatments noted above may also be used. RBV interruption (with or without PEG-IFN) is required within 7 days of stopping TVR if the grade 3 rash does not improve, or sooner if it worsens. If there is any reasonable suspicion or diagnosis of serious cutaneous reactions, or if a skin rash is considered potentially life-threatening, all treatment must be discontinued immediately and permanently.113
Pruritus in the absence of rash is also more common with TVR therapy than with PR49,54 and rarely leads to dose reduction or premature treatment discontinuation. Pruritus can be managed with moisturizing creams, topical cleansing regimens, topical corticosteroids, and/or systemic antihistaminic drugs.
- •
Recommendations:
- °
Close and early monitoring of any skin lesion is needed, especially during TVR treatment (Class 1, Level C).
- °
Management of mild to moderate rash may include emollients, moisturizers, topical corticosteroids, and/or topical or systemic antihistaminic drugs (Class 1, Level C). Management of severe or progressive rash or the presence of a severe cutaneous adverse reaction requires immediate discontinuation of triple therapy, dermatologic consultation, and in some cases emergency hospitalization (Class 1, Level C).
- °
In phase II and III trials, endorectal symptoms were seen more frequently in TVR-treated patients than in controls: 26.2 vs. 5.4%, respectively.49,54 These symptoms included hemorrhoids, anal pruritus, and anal discomfort or rectal burning. They usually occurred within the first 2 weeks of treatment and were mild to moderate; few symptoms required treatment discontinuation, and all resolved after the com- pletion of TVR dosing. The mechanism responsible for these symptoms is unknown but does not appear to be related to other dermatological events. Treatment should be symptomatic with supportive local therapies such as nonspecific topical treatments with or without a local anesthetic in cases of rectal burning. Topical corticosteroids and any allowed systemic antihistamine drugs may also be used for the treatment of pruritus (Class 2a, Level C).
DysgeusiaAmong treatment-naïve patients treated with BOC in phase II and III trials, 35% had dysgeusia, compared with 16% of those treated with PR. The rates of dysgeusia in the treatment-experienced patients were 44% with BOC vs. 11% with PR.9,10,49 Dysgeusia is not a major dose-limiting AE. It has been described as a metallic taste in the mouth and seems to occur throughout the entire treatment. This condition adds discomfort and may lead to reduced appetite and significant weight loss. There is no specific treatment, and some experts recommend chewing gum or drinking chocolate milk when taking the medication114(Class 2a, Level C).
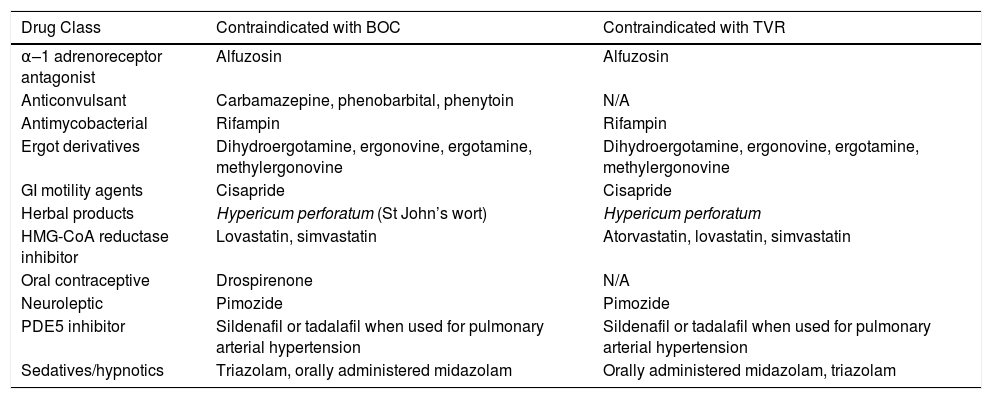
Drug-drug interactionsBOC and TVR are substrates (partly metabolized) and strong inhibitors of CYP3A, so there is a potential for a significant number of drug interactions. BOC and TVR are contraindicated when coadministered with drugs that are potent CYP3A (Cytochrome P450 3A) inducers or that are highly dependent on CYP3A for their clearance. In addition to interactions mediated by CYP3A, TPV and BOC are susceptible to membrane transporter-mediated interactions. Both agents are substrates and inhibitors of P-glycoprotein.86 Several commonly used medications are contraindicated for use with these combination therapies10,113 (Table 8). Other potential interactions can be found using current databases, web pages (http://www.hep-druginteractions.org/), or applications for smart phones or tablets.
- •
Recommendations:
- °
Careful analysis of all prescribed and over-the-counter concomitant medications is needed (Class 2a, Level C).
- °
Every drug used during triple therapy should be monitored (Class 2a, Level C).
- °
Contraindications to BOC and TVR as listed in prescribing information.
| Drug Class | Contraindicated with BOC | Contraindicated with TVR |
|---|---|---|
| α–1 adrenoreceptor antagonist | Alfuzosin | Alfuzosin |
| Anticonvulsant | Carbamazepine, phenobarbital, phenytoin | N/A |
| Antimycobacterial | Rifampin | Rifampin |
| Ergot derivatives | Dihydroergotamine, ergonovine, ergotamine, methylergonovine | Dihydroergotamine, ergonovine, ergotamine, methylergonovine |
| GI motility agents | Cisapride | Cisapride |
| Herbal products | Hypericum perforatum (St John’s wort) | Hypericum perforatum |
| HMG-CoA reductase inhibitor | Lovastatin, simvastatin | Atorvastatin, lovastatin, simvastatin |
| Oral contraceptive | Drospirenone | N/A |
| Neuroleptic | Pimozide | Pimozide |
| PDE5 inhibitor | Sildenafil or tadalafil when used for pulmonary arterial hypertension | Sildenafil or tadalafil when used for pulmonary arterial hypertension |
| Sedatives/hypnotics | Triazolam, orally administered midazolam | Orally administered midazolam, triazolam |
The US Centers for Disease Control and Prevention (CDC) define exposure as an event that might place the health care worker (HCW) at risk of infection. Examples include percutaneous injury (e.g., needlestick or cut with a sharp object) or contact of a mucous membrane or nonintact skin (e.g., exposed skin that is chapped, abraded or afflicted with dermatitis) with blood, tissue, or other body fluid that is potentially infectious (e.g., semen; vaginal secretions; or cerebrospinal, synovial, pleural, peritoneal, pericardial, and amniotic fluids). The risk of infection from these fluids is unknown. Feces, nasal secretions, saliva, sputum, sweat, tears, urine, and vomit are not considered infectious unless they contain visible blood.115 The definition does not include the risk of transmission from performing exposureprone procedures, defined as “those where there is a risk that injury to the worker may result in exposure of the patient’s open tissues to the blood of the worker.” Reitsma, et al. define risk categories and evidence for decision-making in the management of an HCW infected with HCV.116
Risk Factors for Infection with HCV in Health Care SettingsThe US Occupational Health and Safety Administration estimates that 5.6 million persons are at risk of occupational exposure to blood-borne pathogens, and the US National Institute for Occupational Health and Safety reports that 600,000 to 800,000 needlestick and other percutaneous injuries occur annually in the USA,117 leading to around 16,000 new cases of HCV yearly.118 The risk of infection and the associated odds ratio (OR) are as follows: intravenous drug use, OR 49.6; transfusion of infected blood products, OR 10.9 (especially if it occurred before 1992 or 1987 for coagulation concentrates); more than 20 sexual contacts, OR 4.5; sex with an intravenous drug user, OR 6.3; being in prison for > 3 years, OR 2.9; occupational exposure, OR 2.1; piercings or tattoos, OR 2; intravenous immunoglobulin use, OR 1.6; and vertical transmission from mother to child.119 The risk of occupational exposure applies especially to health care workers (HCWs), for whom the estimated prevalence of HCV is 0.28–7.9%.120 The rate of seroconversion after a percutaneous exposure to HCV is 1.8–2.2%;14,15 the highest risk is associated with hollow-bore suture needles, catheterization of a patient’s artery or vein, deep injury, and viremia detected by PCR.121,122 The groups accounting for the higher number of exposure events are nurses (41%), followed by physicians (31%); this difference is partly explained by the greater number of nurses employed in hospitals.123
Strategies to Prevent Transmission of HCV in Health Care SettingsBecause of the lack of effective preexposure and postexposure prophylaxis regimens, prevention is the cornerstone of preventing HCV infection. The first step is to identify the patients at risk. The CDC recommends screening for people who: are current or former injecting drug users (including those who injected once or a few times many years ago); received clotting factor concentrates produced before 1987; were ever on long-term hemodialysis, with persistently abnormal ALT level; were notified that they received blood from a donor who later tested positive for HCV infection; received a transfusion of blood, blood components, or an organ transplant before July 1992; are HCWs and emergency medical and public safety workers who have experienced needlestick or sharps injury or mucosal exposure to HCV-positive blood; are children born to HCV-positive women; or are adults born in 1945–1965, who should be tested once without prior assessment of HCV risk factors.124,125 The latter recommendation has generated a great deal of controversy, especially in developing countries where the data are limited. Nevertheless, studies have shown a five times higher prevalence of HCV infection in this cohort and a slightly higher prevalence in Mexican-Americans.139 This could justify, at minimum, conducting studies in Latin America to determine the validity of the recommendation in this population.
Key measures for minimizing the risk of infection of HCWs with HCV, the so-called ‘standard precautions’, include hand hygiene, personal protective equipment (gloves, gown, mask, goggles, and face shield), soiled-patient care equipment, environmental control, good injection practices, respiratory hy- giene/cough etiquette, etc. These measures should be complemented with administrative, financial, and educational programs directed at every level, and engineering controls (e.g., needleless intravenous medication systems, blunted suture needles, leak-proof secondary containers for transporting blood, and impervious needle disposal containers).
There are several guidelines for the management of an HCV-exposure event, but every institution is responsible for its preventive measures. Figure 5 shows a suggested algorithm for the management and classification of an exposure event. General measures include recording: the name and identification of the source; time, date, and nature of exposure (nonintact skin, mucosal, or percutaneous exposure); the type of fluid, body location, and contact time; the infective status of the source (include date of test); and a description of the injury (e.g., depth of wound, solid vs. hollow needle).

Algorithm for the management of a suspected exposure to HCV, adapted from data in references 124, 126 and 127. HBV: hepatitis B virus. HCW: health care worker. HIV: human immunodeficiency virus. LFTs: liver function tests. PCR: polymerase chain reaction. RNA: ribonucleic acid. EIA: enzyme immunoassay.
The following measures are based on recommendations by the CDC and the US National Institutes of Health:124
- •
Baseline testing for anti-HCV (enzyme immunoassay) and/or HCV RNA by PCR (recommended), and serum aminotransferase (ALT and AST) activity.
- •
Follow-up testing for HCV RNA 4–6 weeks after exposure, and serum aminotransferase activity (ALT and AST) at week 4.
- •
Follow-up testing for anti-HCV (enzyme immunoassay), HCV RNA by PCR, and serum aminotransferase activity (ALT and AST) 3–6 months after exposure.
Current policies regarding HCV-positive HCWs are outdated, and there are no specific restrictions for HCV-positive HCWs, although HCWs known to be HCV positive should comply with their institutional regulations, ensure adequate follow-up, and (if possible) avoid exposure-prone procedures.47
- •
Recommendations:
- °
Patients with risk factors (as defined by the CDC) should be screened routinely for HCV infection (strong recommendation, high quality of evidence) (Class 1, Level A).
- °
HCWs should be screened routinely for anti-HCV. HCV RNA-positive HCWs should avoid exposure-prone procedures (weak recommendation, low quality of evidence) (Class 2b, Level C).
- °
Medical personnel should use protective gear during contact with tissues from HCV-ex-posed patients (Class 1, Level A).
- °
Health care personnel should be educated about protection, good injection techniques, and overall precautions when in contact with a patient’s blood (Class 1, Level A).
- °
Everyone born in 1945–1965 should be tested once for HCV without prior assessment of HCV risk factors (Class 1, Level, B).
- °
In cases of exposure to a patient’s blood products, the exposure site must be washed with soap and water, and samples for hepatitis B virus, HCV, and human immunodeficiency virus (HIV) screening should be taken from both the HCW and patient (Class 1, Level A).
- °
Chronic infection with HCV is an epidemiological problem because it is a chronic infection and because of the risk of dissemination to others.7,47
The prevalence of HCV infection in pregnant women has not been determined exactly. Given the prevalence of infection among all females, one might expect that the prevalence for pregnant women is similar to that of women of similar age within the population. Specific analysis in high-risk groups, intravenous drug users, and women coinfected with HIV has shown that the incidence of infection can be as high as 51%.128,129
The main problem related to HCV infection during pregnancy is mother-to-child transmission, which may occur in 10% of cases. Factors related to such transmission are the mother’s viral load and coinfection with HIV, although breastfeeding and mode of delivery have not been demonstrated to be related to this risk.128,129 In relation to treatment, RBV is teratogenic and cannot be used at any time during pregnancy. There are no reports on the use of IFN during pregnancy.128,129
- •
Recommendations:
- °
HCV infection must be ruled out in all women who have risk factors for HCV infection. Special effort should be taken to identify pregnant women who are intravenous drug users or infected with HIV (Class 1, Level A).
- °
Treatment for HCV during pregnancy must be avoided (Class 1, Level A).
- °
Patients with chronic renal failure are at risk of acquiring HCV because of their greater need for interventional treatments (mainly dialysis, invasive procedures, and transfusions). The prevalence of HCV infection in these patients is variable and is around 20% in developed countries and 10–70% in developing countries.130,131
Because of the adverse long-term impact of HCV infection after kidney transplantation and the current lack of treatment options for HCV after kidney transplantation, treatment should be considered for patients undergoing dialysis. There is evidence that 50% of hemodialysis patients treated with PEG-IFN monotherapy may obtain an SVR, although the therapy may need to be stopped for as many as 23% of patients. Clearance of RBV is through the kidneys, and its use can be dangerous in inexperienced centers. Treatment of patients undergoing hemodialysis may be feasible in units with great experience by using 200 mg every other day.132,133
At present, there is no evidence suggesting that triple therapy should be used in patients with renal failure.
- •
Recommendations:
- °
Patients on dialysis can be treated safely with PEG-IFN monotherapy (Class 2, Level A).
- °
Combination treatment with individualized doses of RBV may be considered in selected patients (Class 2, Level C).
- °
Patients with HCV and end-stage renal disease scheduled for kidney transplantation should undergo antiviral therapy before kidney transplantation (Class 2, Level B).
- °
Progression of liver disease is accelerated in patients with HIV-HCV coinfection, in particular those with a low CD4 (Cluster of differentiation)-positive cell count and impaired immune function, and early antiretroviral therapy should be considered in these patients. If the patient has severe immunodeficiency (CD4-positive cell count < 200 cells/L), active antiretroviral therapy must be used before HCV treatment.134
During PR treatment, didanosine is contraindicated, and stavudine and zidovudine should be avoided; the role of abacavir is undefined. The severity of liver disease must be assessed before beginning therapy, as for patients without HIV infection.135,136 Two phase II trials have demonstrated the usefulness of triple therapy with PIs in patients with HIV-HCV coinfection. One study using BOC and TVR showed significant improvement in the SVR rate.137
- •
Recommendations:
- °
Indications for HCV treatment in HIV-HCV coinfected patients are the same as those in patients with HCV monoinfection (Class 2, Level B).
- °
The same PEG-IFN regimen should be used in HIV-HCV coinfected patients, but RBV should always be dosed according to body weight (Class 2, Level B2).
- °
Triple therapy for HCV in the context of HIV coinfection must be considered in patients with a high risk of progression of liver fibrosis under strict follow-up in centers with experience treating this type of patient (Class 2, level B).
- °
The first analysis of the cost-effectiveness of the PIs in the USA was published recently by Liu, et al.138 Using a decision-analytic Markov model, they evaluated different therapeutic strategies defined according to the use of IL28B genotyping and the type of therapy: standard treatment (PR); triple therapy (standard therapy + a PI); universal triple therapy without considering IL28B polymorphism; and IL28B polymorphism-guided triple therapy that allocated patients with the favorable genotype (CC) to standard treatment and those with the CT/TT genotype to triple therapy. All patients were classified according to the degree of fibrosis (mild vs. advanced).
The approximate cost of weekly treatment with BOC and TVR was calculated as US$1,100 and $4,100 respectively. Because of differences in the approved therapeutic schemes for both therapies, it was estimated that a short treatment course of a general PI would cost $26,400 (28 weeks of treatment with BOC) and that a long therapy course would cost $35,200 (32 weeks of BOC). In the scenario analyses, the PI cost during the first 12 weeks of treatment increased to $49,200 (12 weeks of TVR cost).
This study found that universal triple therapy in patients with advanced fibrosis (assuming an SVR rate with triple therapy of 51 vs. 32% with the standard treatment) decreased the lifetime risk for decompensated cirrhosis from 23.0 to 16.5%, development of HCC from 13.2 to 9.5%, and need for liver transplantation from 4.6 to 3.3%. In patients with mild fibrosis, triple therapy reduced the lifetime risk of decompensated cirrhosis from 8.4 to 5.1%, HCC from 4.7 to 2.9% (assuming an SVR of 61 vs. 38% with the standard treatment), and liver transplantation from 1.5 to 0.9%.109 By contrast, use of IL28B genotype-guided therapy decreased this benefit to 83% compared with universal therapy.
The quality-adjusted life expectancy [measured as quality-adjusted life years or QALYs (Quality-adjusted life year)] increased by 0.54 and 0.67 years in those with advance fibrosis by applying IL28B genotype-guided therapy or universal treatment, respectively, compared with standard treatment. By contrast, these results were inferior (increased by 0.27 and 0.33 years, spectively) for patients with mild fibrosis.
If the cost of triple therapy is $1,100 per week, then universal therapy definitively improves the outcomes but also increases the costs. However, in general, the strategy of universal triple treatment provides more benefit per dollar spent compared with the standard therapy in patients with more advanced fibrosis than in patients with less severe fibrosis. The incremental cost-effectiveness ratio is < US$50,000 per QALY for patients with F4 fibrosis compared with > $150,000 per QALY for those without fibrosis (with a willingness-to-pay threshold of US$50,000). This paper concluded that triple therapy is cost effective when the least expensive PI is used in patients with advanced fibrosis, and that the cost-effectiveness ratio is acceptable regardless of whether universal triple treatment or IL28B genotype-guided therapy is used.123,124 We note that the results of this model were dependent of the cost of PIs, treatment adherence rates, and fibrosis stage.
Another recently published study explored the cost-effectiveness of triple therapy in Europe.139 They used a decision-analytic Markov model and included as the base case a hypothetical cohort of HCV GT1-infected Caucasian, 50-year-old men weighing 70 kg on average who were naïve to treatment and had F2 fibrosis (according to the METAVIR score). Five therapeutic strategies were evaluated: BOC RGT, BOC IL28B genotype-guided therapy, BOC RVR-guided therapy, TVR RGT, and TVR IL28B genotype-guided therapy. They analyzed the shortterm (SVR) and long-term (20 years of follow-up) scenarios. Applying this specific model, the authors found that using the first-generation PIs with the regimens of BOC RVR-guided therapy and TVR IL28B genotype-guided therapy improved survival (life-years gained) by about 4 years and the quality-adjusted life expectancy by about 7 QALYs, by increasing the SVR rate by about 25% with an acceptable incremental cost-effectiveness ratio per QALY of < 10,000 euros (willingness-to-pay threshold of 25,000 euros per life-year gained).125 In contrast to the US study (109), which used universal triple therapy, the implementation of dual PI-free treatment in patients with the IL28B CC genotype or in those who achieved an RVR should have avoided the need for PIs in 25–33% of naïve patients, with a consequent reduction in costs and risks and an improvement in benefits. Nevertheless, the real cost of using one of the PIs in the dual treatment in Europe is estimated at ≥ 2,000 euros, similar to the cost of some oncological therapies.
In separate studies, the cost-effectiveness of BOC and TVR has been evaluated in comparison with the standard of care, and these studies have included both treatment-naïve patients and those who experienced previous therapy failure. Unfortunately, these ana-lyses have been published only in abstract form, and the specific details of the models, as sumptions, case base, and robustness of the data are lacking.
One should keep in mind that cost-effectiveness analyses should not be used to definitively limit or deny therapy to a specific patient. Each country should structure its own model that includes all the local variables (benefits, costs, economic restrictions, etc.) to evaluate new treatment options and to optimize the resources in our cost-constrained countries. Some limited raw data analysis show differences between countries (Table 9).
- •
Recommendations:
- °
Triple therapy (PR + PI) is cost effective in the treatment of chronic HCV GT1-infected patients, especially in those with advanced fibrosis (Class 1, Level A).
- °
Each Latin American country should design a specific model relevant to its own economic conditions, restrictions, and health care systems (Class 3, Level C).
- °
Cost of protease inhibitor treatment according different health system.
| Organization | Boceprevir | Telaprevir |
|---|---|---|
| National Institute for Health and Clinical Excellence (NICE) | • Treatment naïve £ 11,601 | • Treatment naïve £ 13,553 |
| • Treatment experienced £ 2,909 | • Treatment experienced £ 8,688 | |
| Scottish Medicines Consortium (SMC) | • Treatment naïve F0–3 £ 8,800 F4 £ 11,722 | • Treatment naïve All £ 14,230 |
| • Treatment experienced F0–3 £ 7,690 F4 £ 1,368 NR £ 8,042 | • Treatment experienced All £ 9,440 PR £ 5,363 PPR £10,558 NR £ 27,725 | |
| Common Drug Review (CDR) | • Treatment naïve $36,712 | • Treatment naïve $21,901 |
| • Treatment experienced $32,143 | • Treatment experienced PR $1,467 PPR $21,579 NR $36,255 | |
| Pharmaceutical Benefits Advisory Committee (PBAC) | • Treatment naïve Between $15,000-$45,000 | • Treatment naïve Between $15,000-$45,000 |
| • Treatment experienced Between $15,000-$45,000 | • Treatment experienced Between $15,000-$45,000 |
F0-3: non-cirrhotic patients. F4: cirrhotic patients. PR: prior relapser. PPR: previous partial responder. NR: null responder.
After liver transplantation, reinfection of the graft is almost universal. It is important to monitor the progression of fibrosis, which is best monitored by liver biopsy. The presence of significant fibrosis (≥ F2) at 1 year identifies patients who are at risk of losing the graft because of severe HCV recurrence. Such patients are candidates for antiviral treatment. It has also been demonstrated that the presence of portal hypertension (≥ 6 mmHg for F2) 1 year after transplantation identifies with confidence patients at risk of disease progression, with good correspondence to necroinflammation and fibrosis (kappa ≥ 0.6).140 Liver biopsy and measurement of the portal pressure gradient are invasive tests, and if they must be performed repeatedly, as in liver transplantation, they are not well tolerated by the patient. For this reason, noninvasive tests have been combined, such as TE (FibroScan®), which is a fast, easy, and well-tolerated method that measures liver rigidity through a wave propagated through the liver parenchyma. Carrión, et al.141 evaluated prospectively the accuracy of FibroScan in a follow-up study of the severity of HCV recurrence in patients after transplantation. They used repeated measurements using TE to evaluate prospectively 132 patients during the first year after transplantation because of HCV. They performed a liver biopsy and measured the portal pressure gradient 1 year after transplantation. They found that TE was useful for identifying patients with significant fibrosis (F2) or portal hypertension ≥ 6 mmHg, with a cutoff point of 8.5 kPa. The progression speed (kPa/month) was significantly greater in fast fibrosers (0.42 kPa/month) than in slow fibrosers (0.05 kPa/month). The progression speed was also significantly greater in fast fibrosers with cholestasis (1.54 kPa/month).
- •
Recommendations: Despite the utility of TE, liver biopsy is the preferred method to assess fi- brosis after liver transplation in patients with HCV infection (Class 3, Level C).
There is no specific recommendation regarding the perfect timing to perform liver biopsy in these patients (Class 3, Level C).
Future TrendsRecent advances in the understanding of the molecular biology of HCV allow the identification of several targets for antiviral therapy. Some of these drugs have entered phase II and III trials. Preliminary information suggests a very strong antiviral effect with a low incidence of side effects. More time and study results are needed to establish the full implications for clinical practice and the long-term safety of these new drugs.142,143
In addition, several Health Technology Assessment (HTA) agencies around the world had evaluated the triple therapy with both PIs. In Latin America, only Brazil’s HTA Agency (CONITEC: Co-missão Nacional de Incorporação de Tecnologias) had issued recommendation to include both Boce- previr and Telaprevir into the National Health System (SUS: Sistema Único de Saúde). The other body that has issued recommendation to include Boceprevir in the National Formulary is Mexico’s HTA Agency (CSG: Consejo de Salubridad General) (Table 9).
ConclusionIn relation to the previous guidelines, there has been great progress in the therapy of hepatitis C. 144
Despite the advances on therapy for hepatitis C virus infection, according to the Agency for Health- care Research and Quality, the evidence shows an improvement in the management of patients with genotype 1 hepatitis C virus. This information comes for 6 fair-quality clinical trials. There is also an increase in cost and rate of adverse events, nowadays there is no information to take clinical decision considering cost, specific economical conditions or not well represented populations. The algorithm proposed by UK guidelines seems as an non-evidence based, but pragmatic evidence (Figure 6).

Algorithms proposed by UK guidelines for the management of hepatitis C.145
- •
AEs: adverse events.
- •
APRI: aspartate to platelet ratio index.
- •
AST/ALT ratio: aspartate aminotransferase to alanine aminotransferase.
- •
BOC: boceprevir.
- •
CDC: Centers for Disease Control.
- •
CD4: cluster of differentiation.
- •
CNIs: calcineurin inhibitors; inhibit NS3/4A.
- •
CYP3A: cytochrome P450 3A.
- •
DAA: direct-acting antiviral agents.
- •
EPO: erythropoietin-alpha.
- •
eRVR: extended RVR.
- •
FDA: food drug administration.
- •
GT1: genotype 1.
- •
HCC: hepatocellular carcinoma.
- •
HCV: hepatitis C virus.
- •
HCWs: health care workers.
- •
IL28B: interleucina-28B.
- •
OR: odds ratio.
- •
PCR: polymerase chain reaction.
- •
PEG-IFN: pegylated interferon.
- •
PIs: protease inhibitors.
- •
PR: peginterferon and ribavirin.
- •
QALY: quality-adjusted life year.
- •
RBV: ribavirin.
- •
RGT: response-guided therapy.
- •
RVR: rapid virological response.
- •
SVR: sustained virological response.
- •
TE: transient elastography.
- •
TPV: telaprevir.



