



Wilson’s disease (WD) is an uncommon cause of acute liver failure (ALF). Our aim was to describe clinical features, diagnostic findings, treatments, and outcomes of patients with ALF due to WD.
Material and methodsRetrospective medical record reviews of all patients with ALF due to WD in eight years in Uruguay.
ResultsWD was the cause of six (15%) of thirty-nine ALF cases. All patients were females, with a mean age of 18 years. Four patients presented with hyperacute liver failure and two with acute failure. Jaundice was the main complaint of all patients. Mean total bilirubin (TB), alkaline phosphatase (AP), AST, and ALT were 27.5 mg/dL, 45.5 lU/l, 156 IU/L, and 51 IU/L, respectively. Ceruloplasmin levels were low in four patients, urinary cooper was high in four, and two had Kayser-Fleischer rings. All patients had Coombs-negative hemolytic anemia, acute kidney injury, histochemical identifiable copper, and advanced fibrosis on liver histology. The average MELD score was 36. All patients were treated with d-penicillamine and listed for urgent liver transplantation (LT). Prometheus® was performed in one patient. Three patients died: two without LT and one after LT. Three patients survived: one without LT (New Wilson Index<11) and two with LT. The referral time to the program and the total time (referral plus waiting list time) were longer for non-survivors than for survivors (14 vs. 3 days and 23 vs. 8 respectively).
ConclusionAll cases had typical clinical, analytical and histopathology characteristics. Early referral was determinant of prognosis.
Wilson’s disease (WD) has low incidence and prevalence (1 per 30,000 inhabitants),1 and is an uncommon cause of acute liver failure (ALF) (3% of cases in United States,2 6 to 12% in Europe,3 and no cases were reported in Argentina4). It is an autosomal recessive inherited disorder of copper metabolism caused by mutation of the ATP7B gene on chromosome 13. This gene encodes a protein expressed in hepatocytes, which is responsible for transporting copper to the bile (for excretion) and to the blood attached to ceruloplasmin (for its use). The deficit of ATP7B results in reduced biliary copper excretion, reduced incorporation of copper into ceruloplasmin, and the release of free copper into the blood. In consequence, copper accumulates in the liver or other organs such as the brain and cornea.1,5
ALF is an unusual presentation ofWD, accounting only for 5% of all cases.6 It is defined by the presence of coagulopathy with an International Normalized Ratio (INR) > 1.5, plus any degree of encephalopathy, in a patient with an illness of<26 weeks duration, despite having unrecognized cirrhosis in the majority of cases of WD.7 ALF may be the initial clinical presentation of WD or may be a complication in patients who have already been diagnosed and abandoned the treatment. Most of the patients are women in the second decade of life.3,7
There is not a single specific test for WD, so the diagnosis is based on a combination of clinical and analytical criteria:
- 1.
Neurologic symptoms (hypertonia, tremor, dystonia, ataxia) and/or typical abnormalities in brain magnetic resonance imaging (MRI).
- 2.
Decrease of serum ceruloplasmin level (< 20 mg/dL, usually < 10 μ,g/dL).
- 3.
Increase in urinary copper excretion (> 100 ug/24 h = 1, 6 μmol/24 h).
- 4.
Presence of Kayser-Fleischer rings (KF) in the slit-lamp eye exam.
- 5.
Coombs-negative hemolytic anemia.
- 6
ATP7B gene mutation.
- 7
Increased hepatic parenchymal copper concentration (> 250 /xg/g of dry weight of copper in the liver biopsy).3,7
In Uruguay, this last technique is not available, but it is possible to detect copper in hepatocytes by histochemical examination with special stains (orcein and rhodamine).
ALF due to WD (ALF-WD) has a high mortality without emergency liver transplantation (LT). Therefore, establishing a rapid diagnosis is critical for patient management as well as for family screening. However, in the setting of ALF, copper metabolism parameters became less sensitive and specific, while KF rings are only identified in 50% of cases. Most patients have a characteristic pattern of laboratory findings that include: high levels of total bilirubin (TB), normal or subnormal serum alkaline phosphatase (AP) typically<40 IU/L, modest elevation of serum aminotransferases with aspartate aminotransferase (AST) > alanine aminotransferase (ALT), as well as Coombs-negative hemolytic anemia and rapid progression of acute kidney injury (AKI) with altered urinary sediment.3,7,8 Different diagnostic ratios have been considered for quick diagnosis of WD in the setting of ALF. In 1991, Berman described six patients in which the AP/TB ratio < 2 and AST/ALT ratio > 4 had a high sensitivity and specificity for the diagnosis of ALF-WD.9 However, these parameters could not be validated by other reports.10,11 A subsequent study by Korman, et al. in 2008 in a cohort of 16 patients with ALF-WD showed that an AP/TB ratio < 4 had a sensitivity of 94% and a specificity of 96% for diagnosis. In addition, an AST/ALT ratio > 2.2 had a sensitivity of 94% and a specificity of 86%, and the combination of both provided a sensitivity and specificity of 100%.12 Eisenbach, et al.11 showed that the indexes proposed by Berman were not applicable to patients with an average MELD score of 18. Also the AP/TB ratio is not always helpful in children due to the increase of bone AP.13 Therefore, even though these parameters are a guide for diagnosis, they must be used in combination with the conventional diagnostic criteria that we mentioned previously.3
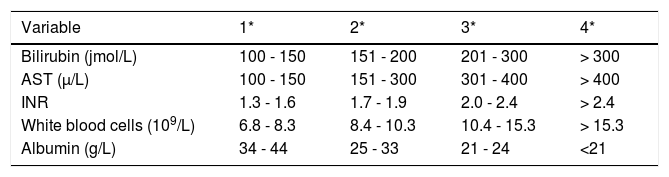
Pharmacological chelation therapy is rarely effective in patients presenting with ALF-WD, mainly due to the time required to remove toxic copper from the organism. However, there are cases of patients surviving without LT, with chelation therapy plus supportive care.11 Plasma exchange and albumin dialysis have been use to rapidly lower serum copper levels in some reports.14,15 A specific prognosis index was developed by Nazer16 in 1986 and subsequently modified by Dhawan, et al.,17 the Wilson Disease Index (WDI). A prognostic score > 11 is associated with a high probability of death without LT (Table 1).3,16
Index forecast in Wilson’s disease, modified by Dhawan, et al.15
| Variable | 1* | 2* | 3* | 4* |
|---|---|---|---|---|
| Bilirubin (jmol/L) | 100 - 150 | 151 - 200 | 201 - 300 | > 300 |
| AST (μ/L) | 100 - 150 | 151 - 300 | 301 - 400 | > 400 |
| INR | 1.3 - 1.6 | 1.7 - 1.9 | 2.0 - 2.4 | > 2.4 |
| White blood cells (109/L) | 6.8 - 8.3 | 8.4 - 10.3 | 10.4 - 15.3 | > 15.3 |
| Albumin (g/L) | 34 - 44 | 25 - 33 | 21 - 24 | <21 |
Uruguay is a country with a population of 3,432,000 habitants18 that has a single Liver Transplant Program, which operates in the Military Hospital, and is an adult only program. Children under 16 years of age are sent to the Hospital Italiano in Buenos Aires-Argentina for LT. The objective of the present study is to describe the clinical features, diagnostic findings, implemented treatments, and the outcomes of patients with ALF-WD in the first eight years of the National Liver Transplant Program in our country.
Material and MethodsWe performed a retrospective, descriptive study. Thirty-nine patients were diagnosis with ALF between April 2009 and April 2017, thirty-three adults and six children. WD was the cause in 6 patients (15%), being the fourth most frequent cause after viral hepatitis, indeterminate cause and autoimmune hepatitis.
WD was diagnosed based on typical symptoms and the presence of conventional biochemical indicators, as previously published.3,7 In all cases, other causes of ALF besides WD were ruled out: consumption of toxic substances was discarded, viral markers and liver autoantibodies were negative, and the abdominal Doppler ultrasound showed no vascular alterations or hepatic infiltration.
Chart report information was collected for study purposes, and a database with the studied variables was created: age, sex, personal and family history of hepatic-biliary-pancreatic diseases, referral time defined by the time between the first consultation and the contact with the LT center, main compliant, time between the installation of the jaundice and the development of encephalopathy to classify as hyperacute, acute, or subacute impairment according to O’Grady’s classification,19 degree of encephalopathy according to the West Haven criteria,20 complementary tests, treatments, time on waiting list for LT, and outcomes.
Institutional Board permission was obtained for reviewing the clinical charts for research purposes.
A statistical analysis was performed to compare referral times, waiting list times and MELD scores between survivors and deceases patients. Wilcoxon rank test was performed with a CI (confident interval) of 95% due to the small size of the sample.
ResultsThe 6 patients were women, with a mean age of 18 years (minimum 12 and maximum 22 years). Only one patient
had a family history of hepatic-biliary-pancreatic disease (one brother died due to acute liver failure, and another brother had an undiagnosed liver disease). Two patients had history of altered liver function, with negative viral biomarkers in the past. The mean referral time to the LT center was 8.5 days (range: 0-15 days).
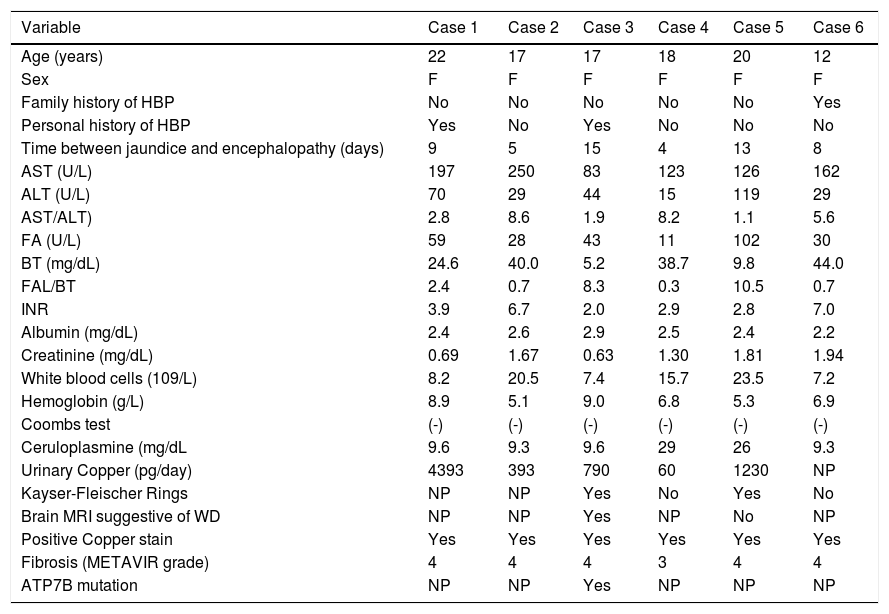
Clinical features and complementary tests (Table 2)Jaundice was the main compliant in all cases. All patients had encephalopathy grade 1 - 2 at the admission to the LT center. Encephalopathy progressed to grade 3 - 4 in three patients. According to the O’Grady classification,18 two cases were hyperacute while the others were acute. The average MELD score was 36 (range: 20 to 49), and five patients had WDI > 11.
Clinical characteristics and complementary test of patients with acute liver failure due to Wilson’s disease.
| Variable | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 |
|---|---|---|---|---|---|---|
| Age (years) | 22 | 17 | 17 | 18 | 20 | 12 |
| Sex | F | F | F | F | F | F |
| Family history of HBP | No | No | No | No | No | Yes |
| Personal history of HBP | Yes | No | Yes | No | No | No |
| Time between jaundice and encephalopathy (days) | 9 | 5 | 15 | 4 | 13 | 8 |
| AST (U/L) | 197 | 250 | 83 | 123 | 126 | 162 |
| ALT (U/L) | 70 | 29 | 44 | 15 | 119 | 29 |
| AST/ALT) | 2.8 | 8.6 | 1.9 | 8.2 | 1.1 | 5.6 |
| FA (U/L) | 59 | 28 | 43 | 11 | 102 | 30 |
| BT (mg/dL) | 24.6 | 40.0 | 5.2 | 38.7 | 9.8 | 44.0 |
| FAL/BT | 2.4 | 0.7 | 8.3 | 0.3 | 10.5 | 0.7 |
| INR | 3.9 | 6.7 | 2.0 | 2.9 | 2.8 | 7.0 |
| Albumin (mg/dL) | 2.4 | 2.6 | 2.9 | 2.5 | 2.4 | 2.2 |
| Creatinine (mg/dL) | 0.69 | 1.67 | 0.63 | 1.30 | 1.81 | 1.94 |
| White blood cells (109/L) | 8.2 | 20.5 | 7.4 | 15.7 | 23.5 | 7.2 |
| Hemoglobin (g/L) | 8.9 | 5.1 | 9.0 | 6.8 | 5.3 | 6.9 |
| Coombs test | (-) | (-) | (-) | (-) | (-) | (-) |
| Ceruloplasmine (mg/dL | 9.6 | 9.3 | 9.6 | 29 | 26 | 9.3 |
| Urinary Copper (pg/day) | 4393 | 393 | 790 | 60 | 1230 | NP |
| Kayser-Fleischer Rings | NP | NP | Yes | No | Yes | No |
| Brain MRI suggestive of WD | NP | NP | Yes | NP | No | NP |
| Positive Copper stain | Yes | Yes | Yes | Yes | Yes | Yes |
| Fibrosis (METAVIR grade) | 4 | 4 | 4 | 3 | 4 | 4 |
| ATP7B mutation | NP | NP | Yes | NP | NP | NP |
- •
Biochemical liver tests.The mean values were: AST 156 (range: 83 - 250), ALT 51 (range: 15 - 119), TB 27.5 (range: 5.2 - 44), AP 45.5 (range: 11 - 102), and INR 4.2 (range: 2 - 7). Four patients had a ratio AP/BT < 4 and AST/ALT > 2.2; the remaining two did not meet any of these criteria.
- •
Hematological profile.All patients had anemia (defined by a concentration of hemoglobin below the expected value based on sex and age), hemolysis parameters (increased LDH), and negative Coombs test. The average hemoglobin was 7.0 g/L (range: 5.1 - 9.0).
- •
Kidney failure (defined as serum creatinine > 1.2 mg/dL) was present in four patients as well as urinary sediment alterations. The rest of them presented kidney failure in the follow-up.
- •
Copper metabolism studies.Four patients had ceruloplasmin levels < 20 mg/dL. Urinary copper was > 100 p-g/day in four of five patients in whom it was determined.
- •
Liver histology.Transyugular liver biopsies were performed in four patients. In one patient, the histology was obtained from the explanted liver and in the rest one from the necropsy. All patients had histochemical identifiable copper and advanced fibrosis.
- •
Slit lamp examination.It was performed in four patients; two of them had KF rings.
- •
Brain magnetic resonance imaging (MRI). It was performed in two cases, one of them showed hyperintense lesions of the deep gray matter at the basal ganglia (Globus pallidus), which are characteristic of copper deposits.
- •
Genetic studies.Determination of ATP7B was performed in only one patient, in which two heterozygote mutations were found in factors c.3207 > A (p.His1069Gin) and c.3809A > G.
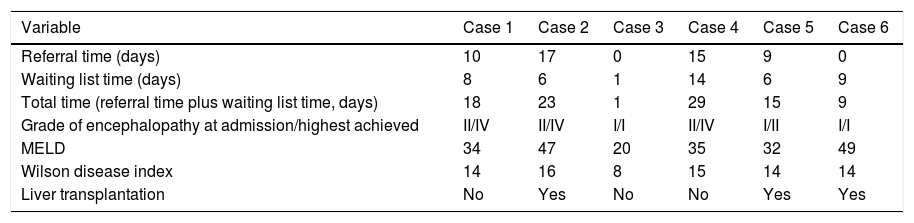
The six patients were treated with d-penicillamine chelation and enlisted for LT. The average waiting list time
Referral time and waiting list time, prognostic indices and outcome.
| Variable | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 |
|---|---|---|---|---|---|---|
| Referral time (days) | 10 | 17 | 0 | 15 | 9 | 0 |
| Waiting list time (days) | 8 | 6 | 1 | 14 | 6 | 9 |
| Total time (referral time plus waiting list time, days) | 18 | 23 | 1 | 29 | 15 | 9 |
| Grade of encephalopathy at admission/highest achieved | II/IV | II/IV | I/I | II/IV | I/II | I/I |
| MELD | 34 | 47 | 20 | 35 | 32 | 49 |
| Wilson disease index | 14 | 16 | 8 | 15 | 14 | 14 |
| Liver transplantation | No | Yes | No | No | Yes | Yes |
was 7 days (range 1 to 14).
Two patients died on waiting list: one due to infection (on the 8th day) and the other due to multiple organ dysfunction (on 14th day). One patient improved and was delisted on the 10th day, being alive at forty-five months of follow-up. This patient had a WDI<11.
LT was performed in three patients. Two adults receive a deceased donor liver transplant at day 1 and 6 on waiting list. One of them survived, being alive at thirty-four months of follow-up, and the other died immediately post-surgery. The child receive a living-donor liver transplant (LDLT) at day 9; artificial liver replacement by method of Prometheus® (Fresenius) as a bridge to the LT was performed. She survived with a follow up of seventy-eight months (Figure 1).
Comparison between survivors and non-survivorsThe referral time to the LT center was significantly higher for the non-survivors than the survivors (14 vs. 3 days, p = 0.04). The mean waiting time on list for nonsurvivors was higher in comparison to survivors (9 vs. 5 days) although there were not statistical significance (p = 0.25). Nevertheless, the total time (referral time plus waiting list time) was significantly longer (23 vs. 8 days, p = 0.05). The mean MELD score was higher in the nonsurvivors than in the survivors (39 vs. 34) although it presented a non-significant p value (0.35). All non-survivors patients reached encephalopaty grade 4, while survivors highest grade of encephalopathy reached was 2.
DiscussionThe diagnosis of ALF-WD is challenging and is based on limited evidence from descriptive studies involving a small number of patients.9-12
Despite the low prevalence of ALF-WD in international series,2-4 in our country WD is the fourth-leading cause of ALF, along with hepatitis B, autoimmune hepatitis, and indeterminate cause.
The clinical presentation was typical: women in the second decade of life, with characteristic biochemical profiles.3,7,8 The four patients who met the diagnostic criteria proposed by Korman12 had the highest MELD scores. All patients met more than one classic diagnostic criterion. Histology obtained from transyugular biopsy confirmed the diagnosis, emphasizing the importance of this procedure in our center.
The mortality was 50%: three of six patients, due to multiorgan dysfunction. Two patients died on waiting list, and one died in the immediate post-LT period. Mortality was associated with the delay in transfer the patients to the LT center. Rivera-Penera, et al. in 1997 previously reported in child patients with ALF that delays in transferring patients to a transplant center significantly affected the likelihood of survival.21 Patients referred late had higher grades of encephalopathy, bilirubin levels, and proportion of subacute evolution,22 which are associated with worst survival.2’4’23 Early referral in patients with ALF may be a key factor in improving patients’ survival because it allows specific and/or highly complex treatment, so it is recommended by international clinical practical guidelines.23
The duration of waiting time on list for LT also can significantly affect patient survival, especially for patients with ALF.24-26 A china study showed that waiting duration was independently correlated with increased mortality with a cut-off value of 5 days for determining the mortality.27 We were not able to show this in our study, potentially due to limitations of the cohort group size, however the total time since the beginning of the symptoms to the outcome (referral plus waiting list time) was associated with mortality.
The two patients who died without LT, waited on list more than 7 days. The two patients transplanted in our center waited less than 7 days. The child patient that received a LDLT was on list more than seven days and survived. However, in this case an artificial liver support system (Prometheus®) was performed. To date, case reports are encouraging that therapeutic plasma exchange and albumin dialysis can either delay or prevent the need for liver transplantation in patients with fulminant hepatic failure due WD. However, these case reports are mainly from the pediatric or young adult population, and thus further studies in adults are warranted.14,15
The patient who survived without LT had the lowest MELD score and a WDI<11, supporting the utility of the score.
In conclusion, all cases of ALF-WD had typical clinical, analytical and histopathology characteristics. Early referral was determinant of prognosis.
Abbreviations- •
AKI: cute kidney injury.
- •
ALF: acute liver failure.
- •
ALT: alanine aminotransferase.
- •
AP: alkaline phosphatase.
- •
AST: aspartate aminotransferase.
- •
INR: international normalized ratio.
- •
KF: Kayser-Fleischer rings.
- •
LT: liver transplantation.
- •
MRI: magnetic resonance imaging.
- •
TB: total bilirubin.
- •
WD: Wilson’s disease.
The authors declares that there is no conflict of interest regarding the publication of this article.
Financial SupportThis study was not supported by grants or external founds.



