



Chromium release assay is the standard method for evaluation of cell-mediated cytotoxicity, including that of mast cells. Although this is a reproducible method, it has more drawbacks than even radioactivity. In addition to the shortcoming of measuring only necrotic killing, some non-radioactive methods have not been widely used either. The numerous limitations of these methods led researchers to develop other techniques. This study describes a new flow cytometric approach that measures human mast cell-mediated cytotoxicity by marking target cells with monoclonal antibody alongside annexin V/ PI co-labelling.
MethodsA colony forming unit - mast in vitro was developed from human bone marrow mononuclear cells in serum-free methylcellulose medium. Six-week-old human bone marrow-derived mast cells were used as effectors, and malignant B-lymphoblastoid cell lines like Daudi / Raji cells as targets. Effectors and targets were both co-incubated for short and long-term durations, and experiments were repeated several times. Cytotoxicity was calculated by flow cytometric mast cell-mediated cytotoxicity assay.
ResultsThis method was able to clearly show mast cell-mediated cytotoxicity against human tumours. It is well-known that some lymphokine-activated killer-sensitive cells are resistant to mast cell-mediated cytotoxicity. However, a different type of lymphokine activated killer-sensitive cell in this study was found to be very sensitive to mast cell-mediated cytotoxicity. Moreover, this technique also allowed us to separate killing into different stages: early and late apoptosis.
ConclusionsWhen compared to chromium release and non-radioactive methods, this method has the advantages of allowing evaluation of early apoptosis and short/long term mast cell-mediated cytotoxicity with specific target marking.
Chromium -51Cr release assay (CRA) has been the standard and most popular method to study cell-mediated cytotoxicity (CMC) including mast cell-mediated cytotoxicity (MCC) in vitro.1. Although this method has the benefits of being reproducible and relatively easy to perform, it has several drawbacks: difficulty in labelling cells with low cytoplasm to nucleus ratios such as Daudi and Raji cells; increased spontaneous release of 51Cr from target cells (≥15%) over time, especially in long-term assays; a delay between actual cell damage and release of 51Cr -bound intracellular proteins into the supernatant; and the inability to measure cytolysis beyond the population level to the single-cell level. Moreover, there is a need for specific care and handling associated with radioactive isotope usage because of the high cost and the short half-life of the radioisotope. Non-radioactive methods have also been used, such as the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [MTT] assay, Hoechst 22147 dye to determine the DNA fragmentation and the electron microscopic evaluation of target cells. In addition to the shortcoming of only being able to measure necrotic killing, these non-radioactive methods also have not been widely used, accepted, or become available.1–3 The numerous limitations of these methods have thus led researchers to measure CMC by other techniques.
A number of groups first developed multicolour flow cytometry (FCM)-based methods using fluorescent membrane and nuclear dyes like DIOC18 and propidium iodide (PI) to study CMC. These methods measure killing by increasing target cell membrane permeability, sufficient to allow access of DNA-binding dyes to the target cell nucleus.1–3 However, the essential element of FCM assays is their ability to discriminate effector cells from target cells by further characterising the fate of the target cell. Recently, several different methods have been suggested to identify target and effector populations, such as the use of differences in light scattering properties. In later studies, researchers utilised monoclonal antibody (mAb) staining for marking target or effector cells in combination with either PI or annexin V (AnnV). AnnV-binding to phosphatidylserine expressed on cells undergoing apoptosis has been successfully used to detect apoptotic cells on FCM.1,2 The addition of AnnV to PI staining further helped with the isolation of different stages of apoptosis taking place in CMC, as demonstrated in our earlier method,1 since PI is retained in cells that had lost their membrane integrity. Thus, the FCM approach seems to have several advantages, including detecting both early and late apoptosis at an individual level with specific target labelling, compared to CRA and non-radioactive methods.
Several years ago, we described a three-colour FCM method to measure CMC1 and called it a “flow cytometric cell-mediated cytotoxicity assay” as well. These established methods have been utilised successfully in our extensive CMC related research.1–11 Based on our earlier experience, the first two-colour FCM assay to elucidate MCC has been recently defined by us.3 The basic strategy of this two-colour FCM assay involves labelling target cells with a fluorescent membrane dye DIOC18, in addition to staining with PI, to identify dead cells. Afterwards, we developed this assay further and used it to determine in vitro human MCC against human tumours. This new three-color FCM approach, called “flow cytometric mast cell-mediated cytotoxicity assay (FCM-MCMCA)” in this article, entails target cell marking with specific mAb (CD19) and with AnnV/PI co-labelling to identify apoptotic/dead target cells. Briefly, a new in vitro human MCC evaluation against human tumours using mAb staining for target cells with AnnV/PI co-labelling on three-colour FCM is described in this study.
Material and MethodsMast (effector) cell development in methylcellulose and maintenance in suspension cultureWe used a modified method to produce a colony forming unit (CFU) -mast in vitro. From several discarded patient samples, human bone marrow mononuclear cells (≥5x104) were obtained and suspended in 0.3ml Iscove's Modified Dulbecco's Medium (IMDM) containing 1% Foetal Bovine Serum (FBS) after Ficoll (Sigma, St. Louis, MO, USA). Cells are put into 3ml serum-free methylcellulose medium [MethoCult™ SFBIT H4236, StemCell Technologies, British Columbia, Canada], supplemented with 200 ng/ml of SCF, 50 ng/ml of IL-6 and 1ng/ml of IL-3 (only at the beginning). All cytokines were purchased from Biosource, Camarillo, CA, USA. We inoculated 0.3ml of the mixed medium in the 12-well plate with a 16 gauge blunt needle and placed it in an incubator. Every two weeks, the cells were fed with 0.3ml of new medium including 100 ng/ml of SCF and 50 ng/ml of IL-6. Thirty or more cells were scored as CFU in situ on an inverted microscope after four weeks. Mast cell (MC) colonies are retrieved from the medium and dissolved with>2-fold volume of phosphate buffer solution including 10% FBS at the fourth week. Cells are centrifuged at 250xG for five minutes. They are suspended and cultured in complete IMDM supplemented with 100 ng/ml of SCF, 50 ng/ml of IL-6, and 2% FBS in a 25cm2 flask up to six weeks for proliferation. The culture was started with ≥104 cells/ml and accumulated up to 2x106 cells/ml. The suspension culture was hemi-depleted every week and supplemented with 100 ng/ml of SCF and 50 ng/ml of IL-6. Some MCs from this study are shown during conjugate formation with tumour cells (Fig. 1A-B).
![A-B. Wright- Giemsa slides showing conjugate formation between mast cell and both tumour cells (effector-target doublets). 1A shows conjugate formation between mast and Daudi cells. 1B depicts conjugate formation between Mast and Raji cells. C-E. Phenotyping of four-week-old human bone marrow-derived mast cells on flow cytometry is shown in a representative sample. 1C shows CD117 (c-kit) expression vs. SS (granularity) of mast cells. 1D demonstrates ≥98% of cells already stained with CD117 and became CD34 negative. 1E illustrates 93% of cells stained with CD33 and 76% of cells were positive for CD49d. [The human effector (mast) cells produced from bone marrow were absolutely negative for CD19 in this study.] 1F-I. Target (Daudi) cell kill, caused by mast cell-mediated cytotoxicity, is demonstrated by flow cytometric mast cell-mediated cytotoxicity assay (FCM-MCMCA) at 12hours in a representative sample. Quadrant gating and determination of populations in (fig. 1F1-I2) were defined by control samples consisting of target/effector alone tubes. The histograms in (fig. 1G2-I2) were achieved from gating accordingly on target cell populations. 1F1 shows location of effector-mast cell in the histograms of FCM-MCMCA. Effector cells are seen as CD19 negative (effector alone sample). 1F2 demonstrates spontaneous kill in effector cell population in control tube. 1G1 shows target-tumour cells location. As expected, target cell population is well-marked with CD19 in the histogram and there is no effector cell (target alone sample) in the tube. 1G2 shows spontaneous kill in target cell population of control sample. It shows 92% viable (Annexin_ / PI-) target (Daudi) cells with total 8% spontaneous killing including early apoptotic plus necrotic kill. 1H1 demonstrates co-incubated effector and target cells at 1:1 ratio after 12 hrs (co-incubation sample). It reveals the changes in the target cell population in the quadrant J of Fig. 1G1 in co-incubated samples after cytotoxic kill. Compared to Fig. 1G1, decrease in size and amount of the target cell population of 1H1 indicates killing due to mast cell–mediated cytotoxicity. Effector cells in this sample are not stained with CD19 and they are in a different area. However; it is hard to say anything about conjugate formation between target and effector cells (effector-target doublets) in the co-incubation sample and it seems to probably be happening earlier. The doublets probably form and immediately dissolve during the beginning of this process. Thus, there is no reflection of conjugate formation in Fig. 1H1. 1H2 histogram of co-incubation sample was acquired after gating on the population in the quadrant J of Fig. 1H1 and reflects the alterations in that co-incubated target population, after cytotoxic kill. It depicts obvious decrease in viability of the target cells from 92% to 48%. It shows increased death up to 52%; 6% of killing was necrotic-late apoptotic (Annexin+/PI+) and 46% of killing was early apoptotic (Annexin+/PI-). Obviously, decline in viability of the target cell population from 92% to 48% strongly reveals significant killing due to human mast cell-mediated cytotoxicity in vitro. Similarly, 1I1 shows further decrease in size and amount of the target cell population of 1G1 and 1H1 due to augmented cytotoxic kill at 2:1 ratio. And figure 1I2 demonstrates decrease in viability of the target cell population from 92% to 37%, but death increased up to 63% at a higher ratio.](https://static.elsevier.es/multimedia/03010546/0000003900000005/v1_201304101054/S0301054610002466/v1_201304101054/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNe5umqYt0zKZP4SYniTZ7+SgZrkCgayp3MRA1ovxa2vU0Y2Yt4vAU6X97OuGC+ayIzgfP3xp0BIjzJY44YHZH5sBVm+hsxNp3iz/ZXPYpdncpLQdCKgdqpcmQqZIXBv4eidptchlQVeg6RVfVR93EeKyjqTaGZYMghMQ4hkOk962YLZTWUtB/ER5tYhC+tLW0zRd++3UA32rY2STomv/agRv6aNjsU7EMIIIK5IzAK8ilGYTIUeUiX6SlsyauymRCjcyicND+xAUZCtocUN1VzH "A-B. Wright- Giemsa slides showing conjugate formation between mast cell and both tumour cells (effector-target doublets). 1A shows conjugate formation between mast and Daudi cells. 1B depicts conjugate formation between Mast and Raji cells. C-E. Phenotyping of four-week-old human bone marrow-derived mast cells on flow cytometry is shown in a representative sample. 1C shows CD117 (c-kit) expression vs. SS (granularity) of mast cells. 1D demonstrates ≥98% of cells already stained with CD117 and became CD34 negative. 1E illustrates 93% of cells stained with CD33 and 76% of cells were positive for CD49d. [The human effector (mast) cells produced from bone marrow were absolutely negative for CD19 in this study.] 1F-I. Target (Daudi) cell kill, caused by mast cell-mediated cytotoxicity, is demonstrated by flow cytometric mast cell-mediated cytotoxicity assay (FCM-MCMCA) at 12hours in a representative sample. Quadrant gating and determination of populations in (fig. 1F1-I2) were defined by control samples consisting of target/effector alone tubes. The histograms in (fig. 1G2-I2) were achieved from gating accordingly on target cell populations. 1F1 shows location of effector-mast cell in the histograms of FCM-MCMCA. Effector cells are seen as CD19 negative (effector alone sample). 1F2 demonstrates spontaneous kill in effector cell population in control tube. 1G1 shows target-tumour cells location. As expected, target cell population is well-marked with CD19 in the histogram and there is no effector cell (target alone sample) in the tube. 1G2 shows spontaneous kill in target cell population of control sample. It shows 92% viable (Annexin_ / PI-) target (Daudi) cells with total 8% spontaneous killing including early apoptotic plus necrotic kill. 1H1 demonstrates co-incubated effector and target cells at 1:1 ratio after 12 hrs (co-incubation sample). It reveals the changes in the target cell population in the quadrant J of Fig. 1G1 in co-incubated samples after cytotoxic kill. Compared to Fig. 1G1, decrease in size and amount of the target cell population of 1H1 indicates killing due to mast cell–mediated cytotoxicity. Effector cells in this sample are not stained with CD19 and they are in a different area. However; it is hard to say anything about conjugate formation between target and effector cells (effector-target doublets) in the co-incubation sample and it seems to probably be happening earlier. The doublets probably form and immediately dissolve during the beginning of this process. Thus, there is no reflection of conjugate formation in Fig. 1H1. 1H2 histogram of co-incubation sample was acquired after gating on the population in the quadrant J of Fig. 1H1 and reflects the alterations in that co-incubated target population, after cytotoxic kill. It depicts obvious decrease in viability of the target cells from 92% to 48%. It shows increased death up to 52%; 6% of killing was necrotic-late apoptotic (Annexin+/PI+) and 46% of killing was early apoptotic (Annexin+/PI-). Obviously, decline in viability of the target cell population from 92% to 48% strongly reveals significant killing due to human mast cell-mediated cytotoxicity in vitro. Similarly, 1I1 shows further decrease in size and amount of the target cell population of 1G1 and 1H1 due to augmented cytotoxic kill at 2:1 ratio. And figure 1I2 demonstrates decrease in viability of the target cell population from 92% to 37%, but death increased up to 63% at a higher ratio.")
A-B. Wright- Giemsa slides showing conjugate formation between mast cell and both tumour cells (effector-target doublets).
1A shows conjugate formation between mast and Daudi cells. 1B depicts conjugate formation between Mast and Raji cells.
C-E. Phenotyping of four-week-old human bone marrow-derived mast cells on flow cytometry is shown in a representative sample.
1C shows CD117 (c-kit) expression vs. SS (granularity) of mast cells. 1D demonstrates ≥98% of cells already stained with CD117 and became CD34 negative. 1E illustrates 93% of cells stained with CD33 and 76% of cells were positive for CD49d. [The human effector (mast) cells produced from bone marrow were absolutely negative for CD19 in this study.]
1F-I. Target (Daudi) cell kill, caused by mast cell-mediated cytotoxicity, is demonstrated by flow cytometric mast cell-mediated cytotoxicity assay (FCM-MCMCA) at 12hours in a representative sample.
Quadrant gating and determination of populations in (fig. 1F1-I2) were defined by control samples consisting of target/effector alone tubes. The histograms in (fig. 1G2-I2) were achieved from gating accordingly on target cell populations. 1F1 shows location of effector-mast cell in the histograms of FCM-MCMCA. Effector cells are seen as CD19 negative (effector alone sample). 1F2 demonstrates spontaneous kill in effector cell population in control tube. 1G1 shows target-tumour cells location. As expected, target cell population is well-marked with CD19 in the histogram and there is no effector cell (target alone sample) in the tube. 1G2 shows spontaneous kill in target cell population of control sample. It shows 92% viable (Annexin_ / PI-) target (Daudi) cells with total 8% spontaneous killing including early apoptotic plus necrotic kill. 1H1 demonstrates co-incubated effector and target cells at 1:1 ratio after 12 hrs (co-incubation sample). It reveals the changes in the target cell population in the quadrant J of Fig. 1G1 in co-incubated samples after cytotoxic kill. Compared to Fig. 1G1, decrease in size and amount of the target cell population of 1H1 indicates killing due to mast cell–mediated cytotoxicity. Effector cells in this sample are not stained with CD19 and they are in a different area. However; it is hard to say anything about conjugate formation between target and effector cells (effector-target doublets) in the co-incubation sample and it seems to probably be happening earlier. The doublets probably form and immediately dissolve during the beginning of this process. Thus, there is no reflection of conjugate formation in Fig. 1H1. 1H2 histogram of co-incubation sample was acquired after gating on the population in the quadrant J of Fig. 1H1 and reflects the alterations in that co-incubated target population, after cytotoxic kill. It depicts obvious decrease in viability of the target cells from 92% to 48%. It shows increased death up to 52%; 6% of killing was necrotic-late apoptotic (Annexin+/PI+) and 46% of killing was early apoptotic (Annexin+/PI-). Obviously, decline in viability of the target cell population from 92% to 48% strongly reveals significant killing due to human mast cell-mediated cytotoxicity in vitro. Similarly, 1I1 shows further decrease in size and amount of the target cell population of 1G1 and 1H1 due to augmented cytotoxic kill at 2:1 ratio. And figure 1I2 demonstrates decrease in viability of the target cell population from 92% to 37%, but death increased up to 63% at a higher ratio.
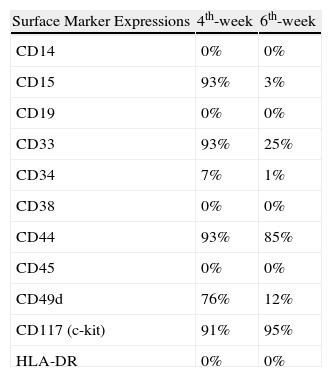
Verification of MCs was done by May-Grunwald-Giemsa, Wright-Giemsa, acid Toluidine Blue staining and immunophenotyping on FCM. In brief, a colony was lifted with an Eppendorf micropipette and spun down at 600rpm for five minutes at the fourth and sixth weeks. Viability was checked with a trypan blue exclusion test. Cells from colonies were stained with May-Grunwald-Giemsa and Wright-Giemsa stains for verification purposes (Figs 1A- B). MCs were fixated with a Carnoy solution and incubated for 2minutes with acid toluidine blue to confirm their tryptase content. MCs were immunophenotyped for all related markers in FCM at the 4th and 6th weeks. CD14, CD15, CD34, CD38, CD45, and HLA-DR markers were detected to be negative. The cells were also negative for CD19 expression. Conversely; CD33, CD44, CD49d, and CD117 (c-kit) expression was positive (Figs 1C- E and Table 1). All mAbs were purchased from Immunotech, Inc. (Westbrook, ME, USA). In this study, although we did not aim to study the mechanisms of MCC; chymase, tryptase, TNF-α and other cytokine expressions were not tested on FCM.
The phenotypic characterisations of 4-to 6-week-old human bone marrow-derived mast cells by flow cytometry.
| Surface Marker Expressions | 4th-week | 6th-week |
| CD14 | 0% | 0% |
| CD15 | 93% | 3% |
| CD19 | 0% | 0% |
| CD33 | 93% | 25% |
| CD34 | 7% | 1% |
| CD38 | 0% | 0% |
| CD44 | 93% | 85% |
| CD45 | 0% | 0% |
| CD49d | 76% | 12% |
| CD117 (c-kit) | 91% | 95% |
| HLA-DR | 0% | 0% |
The human malignant B-lymphoblastoid cell lines, Daudi and Raji, are known to be lymphokine activated killer (LAK)-sensitive and used as the reference cells in CMC studies. All cell lines were obtained from ATCC (Manassas, VA, USA) maintained in RPMI 1640 culture media, and supplemented with 10% FBS. Before co-incubation, target and effector cell viability were determined by the trypan blue exclusion test; a viability of ≥90% was required to proceed. CD19 as a mAb was used successfully for marking both target cells, similar to our earlier CMC studies.1
Flow cytometric mast cell-mediated cytotoxicity (FCM-MCMCA) assay setupFCM-MCMCA was developed from modifications of our earlier established methods known as flow cytometric cell-mediated cytotoxicity assay and two-colour DIOC18/PI method on FCM.1,3 Compared to our earlier two-colour FCM method, a more specific agent (CD19-PE as mAb) was used for target cell marking instead of DIOC18 and AnnV was combined with PI to detect early apoptosis in addition to late apoptotic/dead target cells in this new three-colour FCM approach. Briefly, we performed the following stepwise approach for analysing the samples:
Co-incubationHuman bone marrow-derived MCs at six weeks of age, without any stimulation (PMA etc.), and human tumour targets were co-incubated at certain effector (E) /target (T) ratios (1:1, 2:1, 4:1). Tubes were centrifuged at 115xG for five minutes and incubated at 37°C in 5% CO2 for short and long-term (12 and 24 hrs). Since MCC is well-known to be happening in long-term (>12hrs), 12 hour, and 24 hour co-incubation times were especially selected, as shown in Tables 2A and 2B. In the earlier literature, there has been a tradition of co-incubating murine tumour cells, e.g. WEHI-164, and murine peritoneal MCs for these cytotoxicity assays.12,13 In this study, human bone marrow-derived MCs were co-incubated in vitro with human LAK-sensitive tumour targets for the evaluation of human MCC for short and long-term periods.
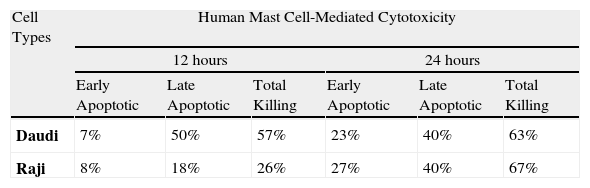
The distribution of mean percentage-based killing in human LAK-sensitive tumour cells at 2:1 ratio on different co-incubation times.
| Cell Types | Human Mast Cell-Mediated Cytotoxicity | |||||
| 12 hours | 24 hours | |||||
| Early Apoptotic | Late Apoptotic | Total Killing | Early Apoptotic | Late Apoptotic | Total Killing | |
| Daudi | 7% | 50% | 57% | 23% | 40% | 63% |
| Raji | 8% | 18% | 26% | 27% | 40% | 67% |
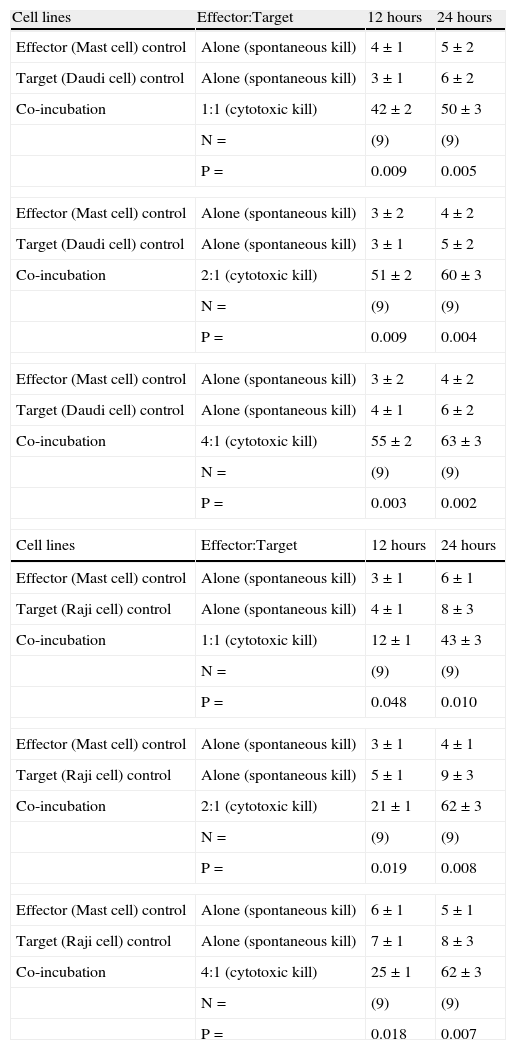
In vitro human mast cell-mediated cytotoxicity % (percentage based killing) measured by FCM-MCMCA against human LAK-sensitive tumour cells at different ratios and time periods (12 hours- 24hours) are shown.
| Cell lines | Effector:Target | 12 hours | 24 hours |
| Effector (Mast cell) control | Alone (spontaneous kill) | 4±1 | 5±2 |
| Target (Daudi cell) control | Alone (spontaneous kill) | 3±1 | 6±2 |
| Co-incubation | 1:1 (cytotoxic kill) | 42±2 | 50±3 |
| N= | (9) | (9) | |
| P= | 0.009 | 0.005 | |
| Effector (Mast cell) control | Alone (spontaneous kill) | 3±2 | 4±2 |
| Target (Daudi cell) control | Alone (spontaneous kill) | 3±1 | 5±2 |
| Co-incubation | 2:1 (cytotoxic kill) | 51±2 | 60±3 |
| N= | (9) | (9) | |
| P= | 0.009 | 0.004 | |
| Effector (Mast cell) control | Alone (spontaneous kill) | 3±2 | 4±2 |
| Target (Daudi cell) control | Alone (spontaneous kill) | 4±1 | 6±2 |
| Co-incubation | 4:1 (cytotoxic kill) | 55±2 | 63±3 |
| N= | (9) | (9) | |
| P= | 0.003 | 0.002 | |
| Cell lines | Effector:Target | 12 hours | 24 hours |
| Effector (Mast cell) control | Alone (spontaneous kill) | 3±1 | 6±1 |
| Target (Raji cell) control | Alone (spontaneous kill) | 4±1 | 8±3 |
| Co-incubation | 1:1 (cytotoxic kill) | 12±1 | 43±3 |
| N= | (9) | (9) | |
| P= | 0.048 | 0.010 | |
| Effector (Mast cell) control | Alone (spontaneous kill) | 3±1 | 4±1 |
| Target (Raji cell) control | Alone (spontaneous kill) | 5±1 | 9±3 |
| Co-incubation | 2:1 (cytotoxic kill) | 21±1 | 62±3 |
| N= | (9) | (9) | |
| P= | 0.019 | 0.008 | |
| Effector (Mast cell) control | Alone (spontaneous kill) | 6±1 | 5±1 |
| Target (Raji cell) control | Alone (spontaneous kill) | 7±1 | 8±3 |
| Co-incubation | 4:1 (cytotoxic kill) | 25±1 | 62±3 |
| N= | (9) | (9) | |
| P= | 0.018 | 0.007 | |
All values are given as mean±SEM. P values reflect the significance of differences between spontaneous and human mast cell-mediated target cell killing. N denotes the number of experiments which were repeated in each series.
This technique is based on labelling the targets after co-incubation with 10μl of mouse anti-human PE conjugated CD19 mAb (Immunotech, Westbrook, ME). Detection of apoptosis/death in target cells was done by staining with AnnV and PI (TACS™ Annexin V-FITC; R&D Systems, Minneapolis, MN, USA) for 20minutes before acquisition. AnnV+ and/or PI+ events were analysed from gating on the events in target population of control as well as co-incubation samples. AnnV+/ PI- events represent an early apoptotic population, and AnnV+/ PI+ dead (necrotic, late apoptotic) cells (Figs 1F2- I2). Thus, this method gives us an advantage of being able to differentiate between late apoptotic/ necrotic cells and early apoptotic cells, compared to CRA.
Flow cytometry acquisition and runningFCM was performed using an EPICS-XL MCL (multicarousel) (Coulter, Miami, FL, USA) equipped with an argon laser (15 mW) source operating at 488nm. The emission of three fluorochromes was recorded through specific band pass filters: 525nm for FITC (FL1), 575nm for PE (FL2), 620nm for fluorospheres (FL3), and 675nm for PI (FL4). The instrument was set for four-colour analysis. As the emission spectra of the three different dyes utilised in this bioassay interfere with one another, appropriate electronic compensations were adjusted by running individual cell populations stained with each dye consecutively through the EPICS. Once the compensations had been set, a gating was done on forward scatter (FS) (ordinate) versus log-scale red fluorescence of mAb CD19 (abscissa) to separate target cells from effector cells (Figs 1F1- I1). The different cell populations were gated with the use of control samples. To measure target-cell death and apoptosis, CD19-positive events were gated on, and analysis of log green fluorescence (AnnV) vs. log red fluorescence (PI) was performed (Figs 1F2- I2).
Flow cytometric data analysisAn average of 10,000 total events and 3,000 target cells were collected per sample. Data were re-evaluated with list mode analysis using Coulter System II™ Version 3 software for the EPICS XL-MCL. The gating for the target cells was based on the target-alone analysis and kept constant throughout all tubes to avoid exaggeration of the counts due to apoptotic body contamination. Cytotoxicity calculations were based on viable populations in target-alone (spontaneous kill in control tube) and co-incubation tube (cytotoxic kill) analysis results. Viable target-cell percentage was determined, and calculations were based on the control-tube values (spontaneous kill). We defined the MCC as percentage cytotoxicity:
Percentage based (cross-sectional) cytotoxicityCytotoxicity calculations were based on viable populations in target-alone and co-incubation tube analysis results, as seen in Table 2A- B. We expressed the MCC as percentage-based (%) cytotoxicity.
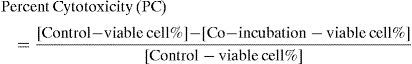
Our in vitro experiments were repeated several times (n=27) with the same set of MC colonies. Samples were tested in duplicates for reproducibility and reliability (mean coefficients of variation (CV) of 1.1% (95% CI, 0.8% -1.7%); r2=0.93, and p<0.001).
Cell staining for microscopy with Wright/GiemsaAt the end of the co-incubation period, the sample was gently mixed and 10μl was used to prepare slides at room temperature without cytospinning. The slides were then stained with Wright/Giemsa. A conjugate formation between tumour cells and MCs was seen (Figs 1A- B).
Comparison with chromium- 51Cr release assay (CRA)A standard assay was performed as previously described.4 Briefly, the assay is a standard 4-hour CRA using target cells that have been pre-labelled with 100μCi 51Cr (Perkin Elmer, Boston, MA, USA) for one hour. Several concentrations of MCs were added to a fixed number of target cells (5,000) in a round-bottom microtiter plate to a total volume of 0.2ml. Following four hours of incubation, 0.1ml of the supernatant was carefully harvested and counted on a scintillation counter (Packard, Downers Grove, IL, USA). Maximum release was determined from wells including target cells and 10% sodium dodecyl sulphate (SDS) in the medium. The PC was calculated using the following equation: (E–S) / (M–S) x100, where E is the experimental counts/minute, S is the spontaneous counts/minute and M is the maximum counts/minute. In the end, when we used our experimental data in comparing FCM-MCMCA with CRA results, there was a significant correlation for PC (n=18; r=0.95; P<0.001), similar to our earlier CMC studies.1
Statistical analysisUnless otherwise specified, all data are presented as means±SEM. The significance of differences between spontaneous kill and cytotoxic kill was determined using the paired Student's t-test and/or Wilcoxon test. The correlation between PC and the CRA results was studied using Pearson's correlation analysis. Significance was considered as<0.05. All statistics were done using the Statistical Package for Social Sciences (SPSS-13 for Windows; Chicago, IL, USA).
ResultsSince human MCs were used instead of murine effector cells, this study has more convincingly demonstrated MCC against human tumour cells, by a better technique, compared to our earlier DIOC18 method.3 Both Daudi and Raji cell death were statistically significant at 12hours as well as 24hours, compared to spontaneous apoptosis/killing in control samples. In vitro human MCC (%) at different ratios and co-incubation times are shown in Table 2B. These findings were consistent with previous murine studies showing murine MCC against lysis-sensitive murine cells, e.g. WEHI-164 and L929.12–14 As shown in these studies, most of the total killing of both human target cells was necrotic kill and it increased over time. Like murine cells, Daudi and Raji are also known to be essentially lysis (TNF-α) –sensitive. Although we did not aim to study the mechanisms, MCC is thought to be happening through pronecrolytic mediators and membranous components of MCs.
In the literature, murine and human MCC did not seem to be very effective against some types of LAK-sensitive cells, such as K562 and YAC-1.14 However, human MCC was found to be very effective against a different type of LAK-sensitive cells, such as Daudi and Raji, as demonstrated in this study.
Moreover, FCM-MCMCA allowed us to separate cytotoxic killing into different stages, early and late apoptotic (necrotic) kill. Distribution of apoptotic type killing according to the cell lines at 12hours was shown at 2:1 ratio in Table 2A. As expected, total killing of both cells increased in due course. Interestingly Raji killing, especially necrotic Raji killing, apparently increased at 24hours, even though Daudi cell killing stayed almost stable. Apoptotic cell death up to 52% was also detected in the samples (Fig. 1H2), indicating the role of pro-apoptotic components of MC granules in MCC. These results seem to confirm the recent literature showing apoptosis induced by MCs in smooth muscle cell, cardiomyocytes and endothelial cells.15,16
Concurrently, short term MCC (≤12hrs) was observed as well, although MCC in the long term is well-known. This is consistent with our earlier results showing short term kill by MCC in DAMI and Meg-01 cells.3,8–11. These results also support the possible contribution of a secretory (exocytosis of pro-apoptotic granules) pathway to MCC that is especially effective in short-term cytotoxicity.
Furthermore, Wright–Giemsa slides showed conjugate formation between MCs and tumour cells (MC-target cell doublets), probably indicating the initial step of MCC with cell-to-cell contact through their membranous components (Fig. 1A-B).
DiscussionIn this study, FCM-MCMCA was demonstrated as having some advantages over DIOC18 and the CRA, e.g. showing early apoptosis with AnnV; better target cell labelling with mAb; and the ability to study long-term MCC as well as identify killing at an individual vs. population level of CRA. Moreover, FCM-MCMCA is also certainly less expensive and specific with mAb marking, without any troubles of radiation and spontaneous release of CRA and dye leakage of DIOC18.1–3
AnnV-binding to phosphatidylserine expressed on cells undergoing apoptosis has been successfully used for a couple of decades to detect apoptotic cells on FCM.1,2,4–8 The addition of AnnV to PI staining further helped with the isolation of different stages of apoptosis, early and late (necrosis) apoptosis. These two states take place in CMC since PI is retained in cells which lost their membrane integrity, as demonstrated in our earlier method.1 Thus, FCM-MCMCA seems to have an upper hand with detecting early as well as late apoptosis at an individual cell level with specific target labelling, compared with CRA and non-radioactive methods such as DIOC18.
While AnnV binds almost irreversibly to the membrane via two C18 alkyl chains, DIOC18 is always questioned with the possibility of dye leakage and consequent labelling of other cells in the environment during co-incubation. In some former studies using target-cell labelling with dyes, cell elimination involving cell membrane disruption or permeabilisation through cytolytic pathways, such as killing by necrolytic granules of MCs, resulted in dye leakage into the medium and artificial staining of other populations in the co-incubation. The use of mAbs does not pose such a disadvantage due to their specificity for either effector or target cells.1,2
An additional aspect of using dyes such as DIOC18 is the need for cell labelling prior to the initiation of co-incubation. Thus, there is also the possibility of affecting the vitality of target and/or cytotoxic activity of the effector cells. Although this may not influence the effector- or target-cell function, this needs to be studied carefully in every new application of this approach. Again, this potential risk is not an issue for our approach due to the addition of mAb at the end of co-incubation, just before using AnnV and PI. Since we did not fixate the cells for mAb staining, surface binding of mAb did not appear to influence cell functioning or staining with other dyes.1,2
The ability to study MCC in longer co-incubation times (≤24 hrs), in addition to shorter co-incubation times (12 hrs), is another advantage of FCM-MCMCA. This appears to be a drawback for CRA, as well as for the methods utilising dyes like DIOC18, due to increased risk in release of 51Cr or dye leakage which results in staining of the other populations. This application is potentially important for studying certain apoptotic pathways that take longer to become operational, such as membrane-bound TNF-α-induced apoptosis, which is believed to be one of the most important components of MCC. For instance, membranous TNF- α- has been shown to kill WEHI-164, L929, and Raji cells in 24 hour assays.12–14,16
More importantly, these results verify the anti-tumour role of MC as a contributory effector cell to other cytotoxic cells in immunosurveillance within human innate immunity. Nevertheless, current literature is very confusing regarding the relation of MCs with tumour cells.17–26 This in vitro study helps elucidate its interactions with tumour cells and may trigger new explanatory studies, such as the delineation of the killing mechanisms of MCC. Contrary to the findings of this study, MC availability in the tumour stroma was assumed to be a stimulator of tumour progression in some recent papers.17–21 Nevertheless, and consistent with this research, increased MC counts were also found to be indicative of a good prognosis in breast, stomach and colorectal cancer in the earlier literature.20–26 Although it is hard to explain these conflicting results, they may be associated with different methodologies, timing of biopsy (performing the biopsy in the early instead of late stage of tumour), the tumour type, and environmental factors surrounding that tumour. Another point is that increased MC density could be a primary and/or secondary result since MC numbers are also found to be physiologically increased around healing tissue, scars, and ovulation. In addition, MCs might be just a reflection of generalised inflammatory reaction of the immune system.20,21 As a result, only observing increased MCs in tumour tissue with good or bad prognosis on pathological specimens is not enough to explain the role of MCs in the tissue, since they are multifunctional, and research results are conflicting. With regard to explaining the relations and role of MCs in tumour cells, further clarification with in vitro and in vivo studies is needed.25,26
In conclusion; FCM-MCMCA convincingly demonstrated human MCC against human tumour targets in this study. Furthermore, FCM-MCMCA is a more definitive, descriptive, and less hazardous approach than our earlier DIOC18 assay and others. However, it is obvious with the known FCM methods that we are still far away from a complete assay which is able to evaluate all components of CMC simultaneously.
Conflict of InterestThe authors have no conflict of interest to declare.
Development of this method was initiated by the author in Wayne State University in Detroit, MI and later completed during his tenure at Louisiana State University–HSC, New Orleans, LA in USA. The author is also very grateful to Liqiao Ma for editing English in the manuscript.
This study was started in Wayne State University and later finished by the author during his time at Louisiana State University - HSC in USA.
