


Common Variable Immunodeficiency (CVID) is characterized by an impaired antibody production and a higher susceptibility to encapsulated bacterial infections. Lung disease is considered to be the most important cause of morbidity and mortality.
MethodsWe analyzed clinical, radiological and functional characteristics in 80 patients with CVID assisted in the Unidad Inmunologia e Histocompatibilidad at Durand Hospital from 1982 to 2018.
ResultsOf the 80 patients, 55 showed pathologic lung Computed Tomography (CT). Twenty of them (36.4%) showed bronchiectasis; 26 (47.3%) interstitial involvement associated with nodules and adenopathies called GLILD (granulomatous-lymphocytic interstitial lung disease); and nine patients (16.3%) showed other lesions. Nine percent of patients with lung disease showed CT progression; none of them had spirometry worsening. GLILD patients had normal and restrictive patterns in lung function tests, in equal proportions. Two patients – one with GLILD and the other one with bronchiectasis – had an increase in spirometric pattern severity without CT progression. Lung biopsy was performed in 19% of GLILD patients, all of whom had histopathologic diagnosis of Lymphoid Interstitial Pneumonia (LIP).
ConclusionsGLILD is the major cause of lung disease in CVID. Computed tomography is useful for diagnosis but not necessary in follow-up, in which functional tests should have better correlation with clinical evolution, reducing radiation exposure. Biopsy should be indicated when the clinical diagnosis is unclear. Treatment should be considered whenever there is clear evidence of disease progression.
Common variable immunodeficiency (CVID) comprises a group of diseases characterized by an impaired antibody production.1 It is the most frequently diagnosed primary immunodeficiency, with an incidence of 1/25,000 to 1/50,000, affecting both genders equally. Symptoms can begin at any age.2 Over 90% of patients suffer from extracellular capsulated bacterial infections, mainly of the respiratory tract. These patients are also more susceptible to autoimmune, granulomatous and oncological diseases, especially stomach cancer and lymphoma.3,4 Diagnosis is based on a significant decrease in serum IgG concentration (two standard deviations below the mean for age) associated with decreased IgA and/or IgM, absence of isohemaglutinins, and/or poor response to vaccines. All of this was analyzed in patients older than 2 years of age, where other causes of humoral immunodeficiency have been excluded.1
Pulmonary involvement due to recurrent infections, lymphoproliferative disease or the combination of both is observed in up to 60% of patients with IDCV. It is one of the main causes of morbidity and accounts for more than half of deaths.5–7 Chest tomographic studies showed bronchiectasis in 20–60% of patients3,8 and, according to different series, interstitial involvement with nodules and adenopathies called GLILD (granulomatous-lymphocytic interstitial lung disease) in 10–32% of patients.3,9–12
GLILD may correspond to granulomas, lymphocytic interstitial pneumonia (LIP), lymphoid hyperplasia or follicular bronchiolitis, which are histological patterns that usually coexist and may be different expressions of the same phenomenon.13 The natural evolution of the disease is unknown, but it could have a progressive course or be the result of various subsequent acute infectious or inflammatory events. There is no conclusive data regarding the effectiveness that the different proposed treatments may have.
Since there is a high morbidity, mortality and incidence of this lung disease in CVID patients, we decided to describe the epidemiological, clinical, radiological and spirometric characteristics in CVID patients assisted in our hospital. This is an effort to establish a strategy for the study of this complication in the early stages of the disease and during its evolution.
Material and methodsAn observational retrospective study was performed. We included all patients with CVID diagnosis assisted at the Unidad Inmunologia e Histocompatibilidad of Dr. Carlos G. Durand Hospital between July 1982 (first patient attended consultation) and July 2018. ESID/PAGID1 criteria were used for the diagnosis of CVID. Immunoglobulin levels were determined by nephelometry or radial immunodiffusion, adopting as the reference those values used by each laboratory.
Epidemiological data was obtained from the medical records; including age, gender, age at symptom onset, and date of first and last visit. The time of evolution for each patient was calculated in years’ time. We considered the age of patients at the beginning of the symptoms and the age they had on their last visit. We analyzed clinical manifestations, tomographic findings, functional lung tests, immunoglobulin values (prior to substitutive treatment), treatment approaches, and the responses to these approaches.
To describe the clinical evolution, we considered as follow-up patients all those who were assisted in our hospital or maintained distant communication, during the 2 months prior to the end of data collection. Deceased patients and those abandoning the follow-up were evaluated up to the time of death or last consultation.
We classified patients with characteristic CVID infections into four phenotypes according to clinical manifestations: (1) infections only; (2) infections and autoimmune disease; (3) infections and lymphoproliferative-granulomatous disease; and (4) infections with autoimmune and lymphoproliferative-granulomatous disease. Autoimmune diseases were classified into two groups: (1) cytopenias and (2) other autoimmune diseases.
Upon clinical suspicion of a respiratory infection, and after performing radiological studies, all patients had sputum tests to look for common and opportunistic germs. Since ours is an endemic region for tuberculosis, in case of suspicion, serial sputum and initiation of empirical tuberculosis treatment were performed.
We divided pneumonia episodes into three groups according to the number of events reported: (1) one to five; (2) six to ten; and (3) 11 or more.
All computed tomography (CT) chest scans performed on each patient were analyzed, and divided into four groups based on the findings: (1) normal pulmonary parenchyma; (2) bronchiectasis only; (3) GLILD; and (4) different abnormalities from those of the previous group. Tomographic studies from patients undergoing acute respiratory infectious processes were excluded. Granulomatous-lymphocytic interstitial lung disease was diagnosed in the presence of solid nodules smaller than 3cm, semisolid nodules, ground glass opacities and thoracic adenopathies.13 In this article we shall call “patients with a diagnosis of GLILD” to all those patients with compatible tomographic findings. Tomographic progression was defined by the occurrence of new lesions or extension of pre-existing ones.
We analyzed the spirometries of all patients, the patterns reported, and their evolution based on the guidelines of the American Thoracic Society.14,15
The frequency of tomographic and spirometric assessment during follow-up was defined by the clinical evolution of each patient.
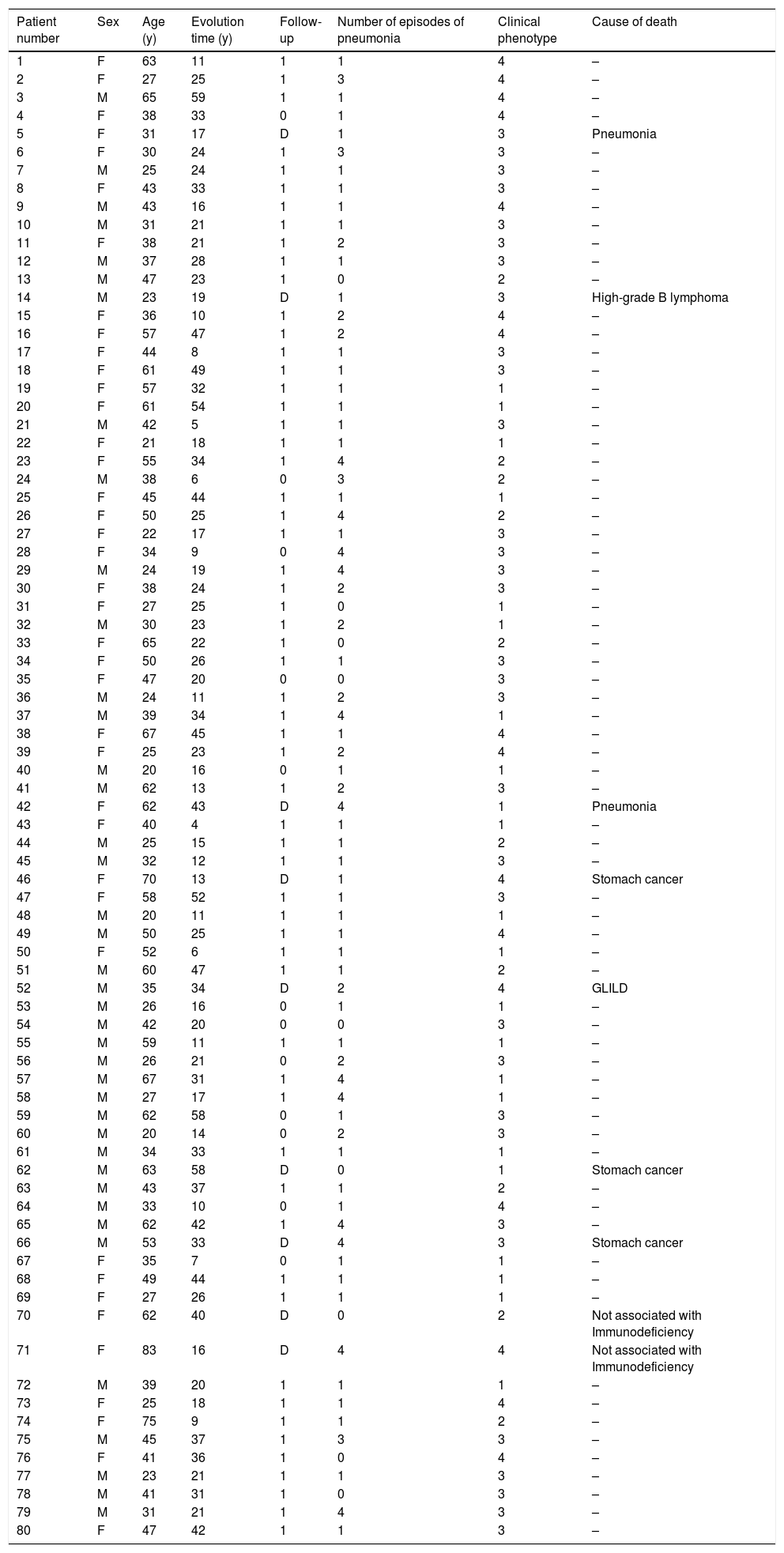
ResultsEighty patients were evaluated: 40 (50%) were women, with a mean age of 42.8±15.4 years at the time of their last visit, and a range from 20 to 83. Fifty-nine patients (73.75%) were in follow-up, 12 (15%) abandoned follow-up, and nine (11.25%) had died at the moment the review was completed. Background patient characteristics are presented in Table 1.
Background patient characteristics.
| Patient number | Sex | Age (y) | Evolution time (y) | Follow-up | Number of episodes of pneumonia | Clinical phenotype | Cause of death |
|---|---|---|---|---|---|---|---|
| 1 | F | 63 | 11 | 1 | 1 | 4 | – |
| 2 | F | 27 | 25 | 1 | 3 | 4 | – |
| 3 | M | 65 | 59 | 1 | 1 | 4 | – |
| 4 | F | 38 | 33 | 0 | 1 | 4 | – |
| 5 | F | 31 | 17 | D | 1 | 3 | Pneumonia |
| 6 | F | 30 | 24 | 1 | 3 | 3 | – |
| 7 | M | 25 | 24 | 1 | 1 | 3 | – |
| 8 | F | 43 | 33 | 1 | 1 | 3 | – |
| 9 | M | 43 | 16 | 1 | 1 | 4 | – |
| 10 | M | 31 | 21 | 1 | 1 | 3 | – |
| 11 | F | 38 | 21 | 1 | 2 | 3 | – |
| 12 | M | 37 | 28 | 1 | 1 | 3 | – |
| 13 | M | 47 | 23 | 1 | 0 | 2 | – |
| 14 | M | 23 | 19 | D | 1 | 3 | High-grade B lymphoma |
| 15 | F | 36 | 10 | 1 | 2 | 4 | – |
| 16 | F | 57 | 47 | 1 | 2 | 4 | – |
| 17 | F | 44 | 8 | 1 | 1 | 3 | – |
| 18 | F | 61 | 49 | 1 | 1 | 3 | – |
| 19 | F | 57 | 32 | 1 | 1 | 1 | – |
| 20 | F | 61 | 54 | 1 | 1 | 1 | – |
| 21 | M | 42 | 5 | 1 | 1 | 3 | – |
| 22 | F | 21 | 18 | 1 | 1 | 1 | – |
| 23 | F | 55 | 34 | 1 | 4 | 2 | – |
| 24 | M | 38 | 6 | 0 | 3 | 2 | – |
| 25 | F | 45 | 44 | 1 | 1 | 1 | – |
| 26 | F | 50 | 25 | 1 | 4 | 2 | – |
| 27 | F | 22 | 17 | 1 | 1 | 3 | – |
| 28 | F | 34 | 9 | 0 | 4 | 3 | – |
| 29 | M | 24 | 19 | 1 | 4 | 3 | – |
| 30 | F | 38 | 24 | 1 | 2 | 3 | – |
| 31 | F | 27 | 25 | 1 | 0 | 1 | – |
| 32 | M | 30 | 23 | 1 | 2 | 1 | – |
| 33 | F | 65 | 22 | 1 | 0 | 2 | – |
| 34 | F | 50 | 26 | 1 | 1 | 3 | – |
| 35 | F | 47 | 20 | 0 | 0 | 3 | – |
| 36 | M | 24 | 11 | 1 | 2 | 3 | – |
| 37 | M | 39 | 34 | 1 | 4 | 1 | – |
| 38 | F | 67 | 45 | 1 | 1 | 4 | – |
| 39 | F | 25 | 23 | 1 | 2 | 4 | – |
| 40 | M | 20 | 16 | 0 | 1 | 1 | – |
| 41 | M | 62 | 13 | 1 | 2 | 3 | – |
| 42 | F | 62 | 43 | D | 4 | 1 | Pneumonia |
| 43 | F | 40 | 4 | 1 | 1 | 1 | – |
| 44 | M | 25 | 15 | 1 | 1 | 2 | – |
| 45 | M | 32 | 12 | 1 | 1 | 3 | – |
| 46 | F | 70 | 13 | D | 1 | 4 | Stomach cancer |
| 47 | F | 58 | 52 | 1 | 1 | 3 | – |
| 48 | M | 20 | 11 | 1 | 1 | 1 | – |
| 49 | M | 50 | 25 | 1 | 1 | 4 | – |
| 50 | F | 52 | 6 | 1 | 1 | 1 | – |
| 51 | M | 60 | 47 | 1 | 1 | 2 | – |
| 52 | M | 35 | 34 | D | 2 | 4 | GLILD |
| 53 | M | 26 | 16 | 0 | 1 | 1 | – |
| 54 | M | 42 | 20 | 0 | 0 | 3 | – |
| 55 | M | 59 | 11 | 1 | 1 | 1 | – |
| 56 | M | 26 | 21 | 0 | 2 | 3 | – |
| 57 | M | 67 | 31 | 1 | 4 | 1 | – |
| 58 | M | 27 | 17 | 1 | 4 | 1 | – |
| 59 | M | 62 | 58 | 0 | 1 | 3 | – |
| 60 | M | 20 | 14 | 0 | 2 | 3 | – |
| 61 | M | 34 | 33 | 1 | 1 | 1 | – |
| 62 | M | 63 | 58 | D | 0 | 1 | Stomach cancer |
| 63 | M | 43 | 37 | 1 | 1 | 2 | – |
| 64 | M | 33 | 10 | 0 | 1 | 4 | – |
| 65 | M | 62 | 42 | 1 | 4 | 3 | – |
| 66 | M | 53 | 33 | D | 4 | 3 | Stomach cancer |
| 67 | F | 35 | 7 | 0 | 1 | 1 | – |
| 68 | F | 49 | 44 | 1 | 1 | 1 | – |
| 69 | F | 27 | 26 | 1 | 1 | 1 | – |
| 70 | F | 62 | 40 | D | 0 | 2 | Not associated with Immunodeficiency |
| 71 | F | 83 | 16 | D | 4 | 4 | Not associated with Immunodeficiency |
| 72 | M | 39 | 20 | 1 | 1 | 1 | – |
| 73 | F | 25 | 18 | 1 | 1 | 4 | – |
| 74 | F | 75 | 9 | 1 | 1 | 2 | – |
| 75 | M | 45 | 37 | 1 | 3 | 3 | – |
| 76 | F | 41 | 36 | 1 | 0 | 4 | – |
| 77 | M | 23 | 21 | 1 | 1 | 3 | – |
| 78 | M | 41 | 31 | 1 | 0 | 3 | – |
| 79 | M | 31 | 21 | 1 | 4 | 3 | – |
| 80 | F | 47 | 42 | 1 | 1 | 3 | – |
M: male; F: female; y: time in years; Follow-up 1: currently in follow-up, 2: abandon follow-up, D: dead; Number of episodes of pneumonia: (1) one to five, (2) six to ten, and (3) 11 or more; Clinical phenotypes 1: (1) infections only, (2) infections and autoimmune disease, (3) infections and lymphoproliferative-granulomatous disease, and (4) infections with autoimmune and lymphoproliferative-granulomatous disease.
All patients had characteristic CVID infections: 22 (27.5%) infections only; 10 (12.5%) autoimmune diseases; 32 (40%) lymphoproliferative-granulomatous disease; and 16 (20%) autoimmune associated with lymphoproliferative-granulomatous disease.
Only nine (11.25%) patients had no history of pneumonia. Of the remaining 71 (88.75%), 44 (61.9%) have had between one to five pneumonias; 11 (15.4%) from six to ten; four (5.6%) 11 or more, and 12 (16.9%) reported multiple episodes without specifying the number.
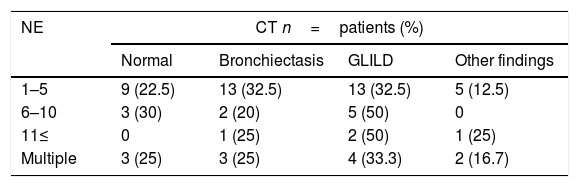
Computed tomography scanSeventy-four (92.5%) patients had chest CT scans. Nineteen of the scans (25.6%) were normal and 55 (74.3%) showed pathological findings. Among them, 20 (36.4%) had bronchiectasis, 26 (47.3%) had images consistent with GLILD, and nine (16.4%) had other lesions. Within this last group, fibrous tracts were diagnosed in six of them: emphysema in one; atelectasis in another one; and isolated interstitial infiltrate in the remaining one. The mean number of CT scans per patient was 6.9±5.2; median five; ranging from one to 30 CT scans.
Relationship between tomographic findings and clinical phenotypeRelationship between tomographic findings and infectionsAmong the 22 patients who presented only characteristic CVID infections, 18 (81.8%) had chest CT scans. Twelve out of these 18 (66.6% of them) had pathological findings: nine had bronchiectasis and three had other findings.
Within the group with no history of pneumonia, eight patients (88.8% of the total) had chest CT scans (four of them were normal; two had GLILD, one bronchiectasis, and the remaining one an isolated interstitial infiltrate).
Thoracic CT scans were performed on 66 (92.9%) out of the 71 patients with history of pneumonia. The diagnosis was GLILD in 24 (36.3%), bronchiectasis in 19 (28.7%), normal findings in 15 (22.7%) and eight of them (12.1%) had other findings (fibrous tracts in six patients, emphysema in one and atelectasis in the other one), see Table 2.
Treatment for tuberculosis was performed in four (5%) patients with GLILD tomographic findings and cervical lymph node involvement. Diagnosis was confirmed in only one of them by means of a bacilloscopy. Two patients improved lymph node involvement with tuberculosis treatment. Lung involvement did not improve in any of them; therefore, alterations were attributed to GLILD. Two other patients with isolated nodal involvement underwent full treatment for suspected nodal tuberculosis, with no microbiological rescue and no improvement by the end of the intervention.
Relationship between tomographic findings and autoimmune, lymphoproliferative and/or granulomatous diseaseA tomography was performed on every patient with an autoimmune disease: alterations were diagnosed in six patients (60%) – two of whom had bronchiectasis and four had other findings.
From 32 patients with lymphoproliferative and/or granulomatous disease, 30 (83.3%) had CT scans. Twenty-four of them (80%) had abnormalities: 16 (66.6%) had GLILD findings, six (24%) had bronchiectasis, and one (4%) had other findings.
All 16 patients with a relationship between lymphoproliferation and autoimmunity had CT scans, 13 (81.25%) of them had pathological findings: 10 with GLILD; two with bronchiectasis; and one with different findings. We diagnosed GLILD in eight out of 12 patients (66.6%) with autoimmune cytopenias and in two out of four (50%) with other autoimmune disease.
Among GLILD patients, nine also had evidence of lymphoproliferative disease in another organ: three in small intestine; three in extra-mediastinal nodes; two in the liver; and one in the muscles, extra-mediastinal nodes and small bowel.
Tomographic developmentThe mean time of evolution was 26.0±10.36 years, range seven to 52 for patients with normal tomography; and 27.13±13.65 years, range six to 59 for those with tomographic alterations. The progression could not be evaluated in 16 of the 55 patients (29.1%) with pulmonary involvement in chest CT due to a lack of comparative studies. Of the remaining 39 (70.9%), 34 (87.1%) did not develop tomographic changes during evolution and five (12.8%) showed progression.
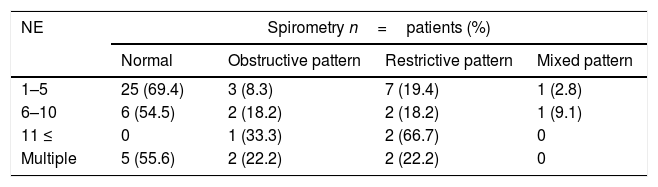
SpirometryAmong the 64 patients who underwent spirometry (80% of the total of 80 patients), 40 (62.5%) had normal findings and 24 (37.5%) were pathological: eight (33.3%) with an obstructive pattern, 13 (54.2%) with a restrictive pattern and three (12.5%) with a mixed pattern. Fifty percent of the patients with an obstructive pattern were severe; 53.8% of those with a restrictive pattern were moderate, and 66.6% of those with a mixed pattern were severe.
Relationship between spirometries and infectionsSpirometry was performed in five (55.5%) out of the nine patients with no history of pneumonia. Four patients (80%) were normal and the remaining patient had a mild mixed pattern.
Spirometry was performed in 59 out of the 71 patients (83.1%) with a history of pneumonia. Thirty-six (61%) had normal results, eight (13.6%) had obstructive patterns, 13 (22%) were restrictive and two (3.4%) were mixed, see Table 3.
Episodes of pneumonia and spirometric patterns relationship.
| NE | Spirometry n=patients (%) | |||
|---|---|---|---|---|
| Normal | Obstructive pattern | Restrictive pattern | Mixed pattern | |
| 1–5 | 25 (69.4) | 3 (8.3) | 7 (19.4) | 1 (2.8) |
| 6–10 | 6 (54.5) | 2 (18.2) | 2 (18.2) | 1 (9.1) |
| 11 ≤ | 0 | 1 (33.3) | 2 (66.7) | 0 |
| Multiple | 5 (55.6) | 2 (22.2) | 2 (22.2) | 0 |
NE: pneumonia episodes.
The mean time of evolution was 26.59±12.28 years, range nine to 54; for patients with normal spirometry and was 26.7±15.86 years, range six to 59 for those with abnormal tests.
It was not possible to assess the evolution in 10 out of the 24 patients (41.7%) with pathological spirometry due to a lack of comparative examinations. Of the remaining 14 (58.3%), 12 (85.7%) maintained stable values during their evolution and two (14.3%) developed progression (one with GLILD findings and the other with bronchiectasis – both without tomographic progression).
Anatomopathological studyFive out of the 26 patients (19.2%) with images suggestive of GLILD had biopsies, and the histopathology was compatible with LIP, describing multifocal interstitial lymphoid infiltrates spreading into the alveolar septa and surrounding airways and vessels. The infiltrates were composed of small lymphocytes admixed with plasma cells. The lymphocytes were a mixture of polyclonal B cells (CD20 positive, mainly in nodules) and T cells (CD3 positive, mainly in pulmonary interstitium). No differences were observed in the spirometric evolution or in the CT scans of these patients.
TreatmentDuring the follow-up in our hospital, only two patients with GLILD were treated for their pulmonary involvement with high-dose corticosteroids. Neither showed improvement. After steroid treatment, one of the patients showed no tomographic or spirometric progression and therefore did not require additional treatment. The other patient was proposed for a lung transplant because of the severity of his condition and his repeated infections. The patient died while waiting for the transplant.
MortalityThe cause of death in three out of nine patients (33.3%) was lung disease (two of them died due to infection and the third one because of the development of the disease). In three of the remaining six patients, the most frequent cause of death was stomach cancer. Another patient developed a high-grade B lymphoma following a history of lymphoproliferative disease, GLILD and extra-mediastinal nodal involvement of more than 15 years of evolution. The cause of death of the remaining two patients was not associated with the immunodeficiency.
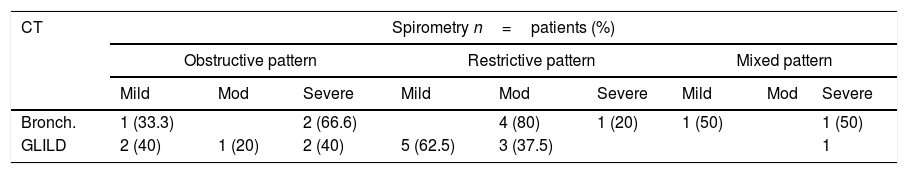
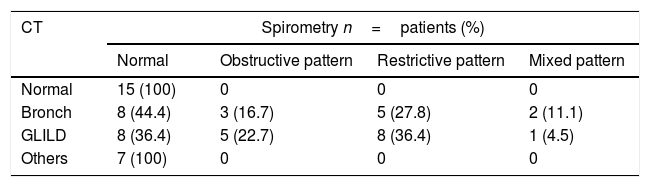
Thorax tomography and spirometry relationLung function was evaluated and found to be normal in 15 of the 19 patients with a normal CT (78.9%). Spirometry was performed in 86.95% of the patients with CT alterations, and 60% of them had alterations. Seven (77.8%) patients with other tomographic findings had normal lung functional testing, as shown in Tables 4 and 5.
Spirometric pattern severity in patients with bronchiectasis and GLILD.
| CT | Spirometry n=patients (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Obstructive pattern | Restrictive pattern | Mixed pattern | |||||||
| Mild | Mod | Severe | Mild | Mod | Severe | Mild | Mod | Severe | |
| Bronch. | 1 (33.3) | 2 (66.6) | 4 (80) | 1 (20) | 1 (50) | 1 (50) | |||
| GLILD | 2 (40) | 1 (20) | 2 (40) | 5 (62.5) | 3 (37.5) | 1 | |||
Bronch.: bronchiectasis; Mod: moderate.
Tomographic findings and spirometric pattern relationship.
| CT | Spirometry n=patients (%) | |||
|---|---|---|---|---|
| Normal | Obstructive pattern | Restrictive pattern | Mixed pattern | |
| Normal | 15 (100) | 0 | 0 | 0 |
| Bronch | 8 (44.4) | 3 (16.7) | 5 (27.8) | 2 (11.1) |
| GLILD | 8 (36.4) | 5 (22.7) | 8 (36.4) | 1 (4.5) |
| Others | 7 (100) | 0 | 0 | 0 |
Bronch.: bronchiectasis.
We describe the epidemiological data, clinical characteristics, and evolution of 80 patients with CVID diagnosis assisted in our hospital unit during the last four decades. Epidemiological characteristics were similar to those reported by other authors.4,5,16
Clinical phenotypesFrequency of clinical phenotypes differs among different series reported by groups from different regions.5,17,18 All the patients in our series presented characteristic infections in contrast with that reported by other groups, where up to 6% do not have characteristic CVID infections.5,17,18 Pneumonia has been reported in 40 to 87% of patients, according to different series, while in ours it was 88.75%, which is slightly higher. As in other groups, most patients had had between one to five episodes of pneumonia.3,5,18–20 Lymphoproliferative-granulomatous disease affected 60% of our patients, including 20% associated with autoimmune diseases. This is significantly greater than the 8–22% reported by other groups, becoming a unique feature of our population.3,5,9–12 The association between autoimmune and lymphoproliferative diseases has been widely described with a variable diagnosis frequency between 8 and 54%.3,12,17
Computed tomography scans and spirometryChest CT scans were performed in over 90% of patients, while spirometries were done in over 80%. The difference observed towards imaging studies may be explained by the fact that these are requested in a mandatory way during acute processes or exacerbations of respiratory infections, while functional respiratory studies used for monitoring depend on patient adherence.
Computed tomography scansTomographic alterations were more frequent in patients with lymphoproliferative-granulomatous disease (80% of cases) and in patients with association between lymphoproliferative-granulomatous and autoimmunity (81.25% of cases), compared to those who only had infections and/or autoimmune diseases (approximately 65% of cases).
Bronchiectasis has been described in 20 to 60% of patients with CVID and has been associated with recurrent infections, delayed immunodeficiency diagnosis, and inappropriate treatment.3,8 However, development of structural airway disease seems to progress despite adequate immunoglobulin replacement and even in the absence of clinically recognized infections. This suggests that there are additional factors that contribute to its development.11,20,21 One of the patients with no history of pneumonia in our series had bronchiectasis, while in patients with a history of pneumonia the frequency of bronchiectasis was greater than 30%. However, we found a difference of less than 5% among groups, according to the number of episodes of pneumonia. This would suggest that infections are not the only etiological factor and that the development of bronchiectasis seems to be independent of the number of infections. This indicates that chest CT at the time of CVID diagnosis would be useful, regardless of a history of respiratory infections.
We found no reports about how the number of infections and the development of GLILD are associated. In our series, we observed that the frequency of GLILD doubles on chest CTs, when patients present more than five episodes of pneumonia. This suggests a relationship between multiple infections and pulmonary lymphoproliferative disease.
The association of GLILD and autoimmunity has been described with a diagnosis frequency between 38% and 50%,12,18 which is lower than what we have found in our series (over 60%). Our patients showed an association between GLILD and autoimmune cytopenias higher than 60%, whereas with other autoimmune diseases it was of 50%. The association between GLILD and cytopenias and between GLILD and other autoimmune manifestations was not found in other series.
GLILD was the only manifestation of lymphoproliferative disease in more than 35% of patients in our series.
SpirometrySpirometry was normal in 80% of patients with no history of pneumonia and in more than 60% of patients with pneumonia. The percentage of patients with alterations in spirometry is greater than 20% in those with a history of pneumonia. However, this value increases considerably, reaching 60% in those patients with more than 10 reported episodes. This suggests a relationship between the number of infectious diseases and the functional respiratory alterations.
HistopathologyThe histopathology reported was compatible with LIP in all our GLILD patients who underwent pulmonary biopsies. In agreement with other specialists, we consider that if biopsies were performed in all patients with GLILD tomographic findings, the most frequent anatomopathological diagnosis would be benign polyclonal lymphoproliferation disease.10,12 From our point of view, lung biopsy should be a procedure indicated only when there is a high clinical suspicion of a disease other than GLILD, such as lymphoma or tuberculosis, when a simple and less invasive diagnostic procedure is not available.
TreatmentDifferent treatments with high-dose corticoids, immunosuppressive drugs, immunomodulators and biological agents have been proposed for patients with GLILD. Their usefulness has been documented in few cases and there are no conclusive studies relating to their effectiveness.6,10,11,13,22–26 Most of our GLILD patients had no clinical tomographical or functional progression which would justify any treatment. Based on our experience, we consider that treatment should be indicated when active disease is evident in GLILD patients, since most patients do not seem to present clinical or spirometric deterioration during the evolution of their disease. Moreover, different immunosuppressive treatments proposed could cause more risks than benefits.
MortalityThe percentage of mortality in our series is 11.25%, whilst in other groups it ranges from 6% to 19.6%.5,21 Among our patients, the main causes of death were pulmonary complications, as reported by other groups, and stomach cancer with a strikingly high incidence.
Thorax tomography and spirometry relationThorax tomography showed alterations over 70% of our patients, with normal functional examination in 40% of them, showing that tomographic abnormalities would have no correlation with functional alterations in a group of patients.
All patients with normal CT scans or with CT findings other than GLILD and bronchiectasis had normal spirometries, suggesting that CT images different from bronchiectasis and GLILD would probably not be of clinical relevance. Fibrous tracts as an isolated finding in tomography would not have an influence in clinical evolution. We found no data describing this relationship.
It has been described that patients with bronchiectasis tend to develop mainly obstructive patterns in spirometry,8,16 whereas patients with GLILD generally have restricting patterns.6 In our series we observed that almost half of the patients with bronchiectasis had normal spirometry, while patients with GLILD presented normal spirometries and restrictive patterns in equal proportions. These data show that a considerable percentage of patients have tomographic alterations without functional alterations. We did not find any relationship between time of evolution and tomographic or spirometric alterations as described by other authors.6,12
A chest CT has been recommended at the time of diagnosis of CVID and during follow-up, with different time intervals (1–5 years) to assess the development or progression of lung disease.12,27 Alternatively, in order to minimize risks of radiation exposure, some authors suggest performing CT scans in 4- to 5-year intervals with annual pulmonary function tests.28 However, there are no studies that justify such recommendations about routine CT scans and the frequency of CT performing in follow-up assessment. Pulmonary function tests are inferior to screening images for early lung disease but are useful in monitoring the progression of chronic lung disease and deciding on therapeutic management.6,13 This observation shows that both tests evaluate different aspects of the same lung disease.7,27
In our series, we observed that only two patients had a progression in spirometry and none of them had tomographic progression. Furthermore, we did not observe worsening in spirometry in any of the patients with tomographic progression, which would indicate that respiratory functional examination, unlike CT, could be useful in the follow-up of patients with pulmonary pathology, thus avoiding the risk of exposure to radiation.
Bronchiectasis and especially pulmonary lymphoproliferative granulomatous disease are the main causes of morbidity and mortality in patients with CVID. Computed tomography is necessary for diagnosis, but has not demonstrated clinical utility during follow-up, where respiratory functional study could have a better correlation with the clinical evolution of patients – without the associated radiation risk. Lung biopsy is an invasive process that would only be indicated when clinical manifestations are highly suggestive of a disease different from GLILD. Treatment in GLILD patients should be indicated when active disease is evident.
Conflict of interestThe authors have no conflict of interest to declare.
