








Among the temporal bone abnormalities that can be found in the etiological study of paediatric sensorineural hearing loss (SNHL) by imaging techniques, those related to the internal auditory canal (IAC) are the least frequent. The most prevalent of these abnormalities that is associated with SNHL is stenotic IAC due to its association with cochlear nerve deficiencies. Less frequent and less concomitant with SNHL is the finding of an enlarged IAC (>8mm).
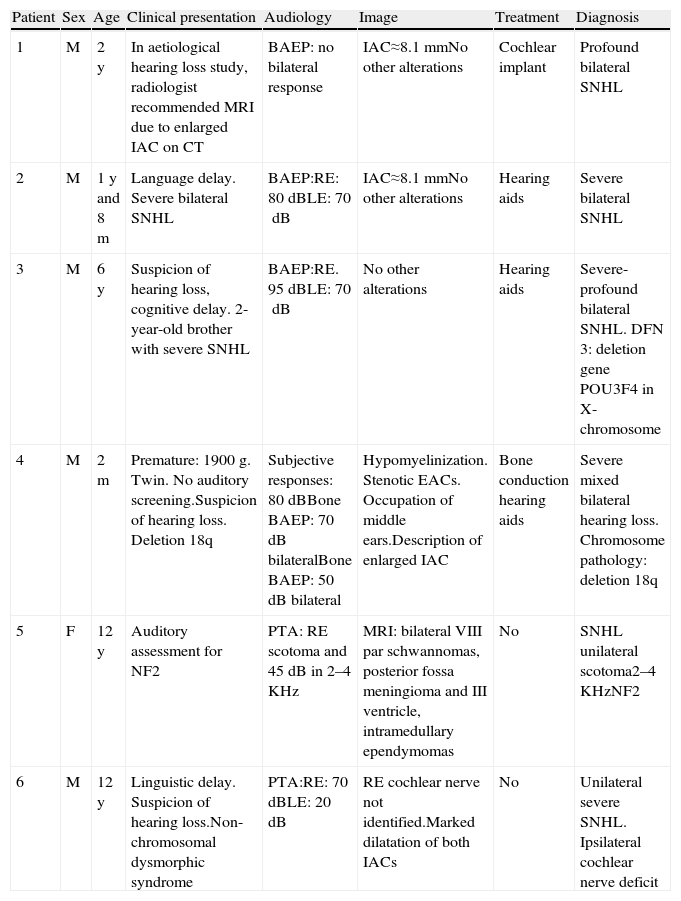
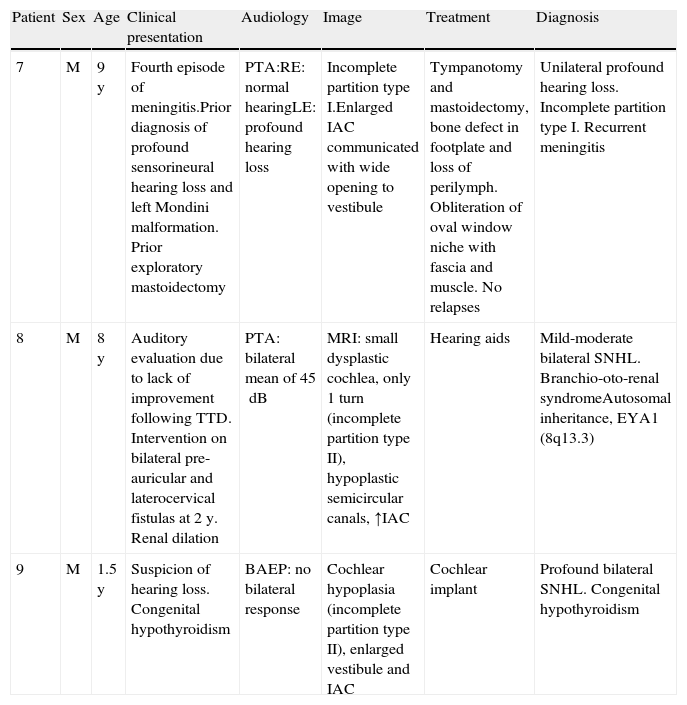
MethodsRetrospective and descriptive review of clinical associations, imaging, audiological patterns and treatment of 9 children with hearing loss and enlarged IAC in the period 1999–2012.
ResultsTwo groups of patients are described. The first, without association with vestibulocochlear dysplasias, consisted of: 2 patients with SNHL without other temporal bone or systemic abnormalities, one with bilateral mixed HL from chromosome 18q deletion, one with a genetic X-linked DFN3 hearing loss, one with unilateral hearing loss in neurofibromatosis type 2 with bilateral acoustic neuroma, and one with unilateral hearing loss with cochlear nerve deficiency. The second group, with association with vestibulocochlear dysplasias, was comprised of: one patient with moderate bilateral mixed hearing loss in branchio-oto-renal syndrome, one with profound unilateral SNHL with recurrent meningitis, and another with profound bilateral SNHL with congenital hypothyroidism.
ConclusionsThe presence of an enlarged IAC in children can be found in different clinical and audiological settings with relevancies that can range from life-threatening situations, such as recurrent meningitis, to isolated hearing loss with no other associations.
Entre las anomalías del hueso temporal que pueden encontrarse en el estudio etiológico de la hipoacusia neurosensorial (HANS) infantil mediante pruebas de imagen, las relacionadas con el conducto auditivo interno (CAI) se hallan entre las menos frecuentes. De ellas, la más prevalente y relacionada con HANS es el CAI estenótico por su asociación a deficiencias del nervio coclear. Menos frecuente y menos concomitante con HANS es el hallazgo de un CAI agrandado (> 8mm).
MétodosEstudio retrospectivo y descriptivo de las asociaciones clínicas, estudios de imagen, patrones audiológicos y opciones de tratamiento de 9 niños diagnosticados de hipoacusia en el periodo 1999–2012 con un CAI agrandado.
ResultadosSe describen 2 grupos de pacientes. El primero, sin asociación con displasias cocleovestibulares: 2 pacientes con HANS sin otras alteraciones de hueso temporal o sistémicas, una hipoacusia mixta bilateral con cromosomopatía por deleción 18q, una hipoacusia genética DFN 3 ligada a X, una hipoacusia unilateral en neurofibromatosis tipo 2 con neurinoma del acústico bilateral, y una hipoacusia unilateral con déficit de nervio coclear unilateral; y un segundo grupo con asociación a displasias cocleovestibulares: una hipoacusia mixta bilateral moderada en síndrome branquio-oto-renal, una HANS profunda unilateral con meningitis recurrentes, y una HANS bilateral profunda con hipotiroidismo congénito.
ConclusionesLa presencia de un CAI agrandado en niños puede encontrarse en diferentes contextos clínicos y audiológicos, con relevancias que pueden variar desde situaciones con riesgo vital como en meningitis recurrentes, hasta hipoacusias aisladas sin otras asociaciones.