








Dengue viral infection affects approximately 130 countries worldwide. According to WHO reports, 40% of the global population lives in rural areas with a high risk of contracting dengue. Researchers have identified four distant strains of the dengue virus, and a single vaccine has not permanently controlled the emergence of all four distant strains. Therefore, a vaccine is required for each of the four strains to address the current situation.
ObjectivesThe objective of this study was to design a multi-epitope-based vaccine for the dengue virus-2 strain that elicits a robust immune response while being safe and non-allergenic.
ResultsFirstly, we analyzed the envelope protein for its physicochemical and antigenic properties. Next, we predicted MHC I, MHC II, and B-cell epitopes with high accuracy and evaluated their properties. Then, we constructed a vaccine using a suitable adjuvant and linkers, and predicted the secondary and tertiary structure of the vaccine, and the tertiary structure was validated. After conducting molecular docking with toll-like receptors, we utilized the best-docked result for molecular stimulation. Finally, we analyzed the immune stimulation against the vaccine, and the results showed positive immune responses from macrophages, DC cells, T-cells, and B-cells. Additionally, we found that the vaccine was excreted from the human body.
ConclusionsOur study demonstrates the potential of using immunoinformatic tools and immunological knowledge to design a multi-epitope-based vaccine for the dengue virus-2 strain. This approach could be applied to designing vaccines for other diseases, and further studies are required to validate its effectiveness in vivo.
La infección viral por dengue afecta aproximadamente a 130 países a nivel mundial. Según informes de la OMS, el 40% de la población mundial vive en zonas rurales con alto riesgo de contraer dengue. Los investigadores han identificado cuatro cepas distantes del virus del dengue y una sola vacuna no ha controlado permanentemente la aparición de las cuatro cepas distantes. Por lo tanto, se requiere una vacuna para cada una de las cuatro cepas para hacer frente a la situación actual.
ObjetivosEl objetivo de este estudio fue diseñar una vacuna basada en múltiples epítopos para la cepa del virus del dengue-2 que provoque una respuesta inmunitaria robusta a la vez que sea segura y no alergénica.
ResultadosEn primer lugar, analizamos la proteína de la envoltura en cuanto a sus propiedades fisicoquímicas y antigénicas. A continuación, predijimos epítopos de MHC I, MHC II y células B con gran precisión y evaluamos sus propiedades. Luego, construimos una vacuna utilizando un adyuvante y enlazadores adecuados, y predijimos la estructura secundaria y terciaria de la vacuna, y se validó la estructura terciaria. Después de realizar el acoplamiento molecular con receptores tipo toll, utilizamos el mejor resultado acoplado para la estimulación molecular. Finalmente, analizamos la estimulación inmunológica contra la vacuna y los resultados mostraron respuestas inmunológicas positivas de macrófagos, células DC, células T y células B. Además, encontramos que la vacuna se excretaba del cuerpo humano.
ConclusionesNuestro estudio demuestra el potencial del uso de herramientas inmunoinformáticas y conocimiento inmunológico para diseñar una vacuna basada en múltiples epítopos para la cepa del virus del dengue-2. Este enfoque podría aplicarse al diseño de vacunas para otras enfermedades, y se requieren más estudios para validar su eficacia in vivo.
Dengue fever is a significant public health issue for individuals living in tropical and sub-tropical regions, where almost one-third of the world's population is at risk of contracting the disease. In recent decades, dengue fever has become increasingly prevalent throughout the world.1 Based on a survey conducted by the World Health Organization (WHO), worldwide approximately 390 million new cases of dengue fever are diagnosed each year. The disease is already prevalent in 130 countries across Asia, the Pacific, America, Africa, and the Caribbean. In India, dengue fever cases have increased fivefold in the past five years, according to the National Vector Borne Disease Control Program (NVBDCP).2,3
Dengue virus (DENV), belonging to the Flaviviridae family, is a single-stranded RNA positive virus with a genome size of approximately 10.7 kb. Researchers have identified four different serotypes of DENV, designated as DEN-1, DEN-2, DEN-3, and DEN-4. Interestingly, these serotypes are genetically closely related, but they differ in terms of their antigenicity.4 The DENV genome comprises of a one polyprotein and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The capsid protein, also known as protein C, plays a crucial role in virus budding, membrane fusion, and nucleocapsid assembly by packaging viral RNA.5 Another protein of the virus, the peptide pr, binds to the envelope protein E in the trans-Golgi at pH 6.0 to prevent early fusion of envelope proteins.6 The prM protein is the only viral protein matured in the trans-Golgi network, and it plays a critical role in masking and inactivating the envelope protein E fusion peptide.7 The envelope protein E is composed of a transmembrane region and an ectodomain, which is further divided into three domains: Envelope domain-1, which is responsible for structural organization; Envelope domain-2, which is responsible for host cell fusion; and Envelope domain-3, which is responsible for viral budding. The envelope protein is responsible for delivering the viral genetic material into the host cell and also serves as an antigen that helps human immune cells recognize and fight against the virus. This envelope protein are important components in vaccine production and are a key target for viral inhibition and neutralization.8 Despite the lack of preventative drug or targeted therapy for dengue fever, adequate intensive care and diligent health administrations are needed to treating the disease. However, vaccines not only provide immunity against pathogens but also aid in controlling vector-borne infectious diseases.9
Although the first dengue vaccine was developed in 1929, numerous vaccines have been tested in clinical trials, but none have proven effective against the DENV.10 A new study shows the evolution of Dengvaxia, a live attenuated tetravalent dengue vaccine that was developed by Sanofi Pasteur. In December 2015, it was first accepted in Mexico to treat people aged 9 to 45 who live in endemic areas. However, due to its recombinant live attenuated nature, there is a high possibility of the attenuated vaccine strain reverting to a more lethal virus strain. Moreover, clinical trials have revealed that Dengvaxia is ineffective against the DENV-2 strain. Many individuals are experiencing severe allergic reactions, such as allergies, urticaria, and headaches, due to ongoing vaccine trials. Additionally, clinical trial results for Dengvaxia indicate that it has harmful effects on pregnant women, resulting in miscarriage, stillbirth, elective termination, and uterine death. This situation highlights the need for the development of a new dengue virus vaccine candidate that can effectively combat the DENV-2 strain.11 Immunoinformatics offers an in-silico approach to develop a reliable and multi-epitope vaccine in a shorter timeframe, cost-effective alternative to the conventional laboratory-based vaccine production method.12 Therefore, in this study, our aim was to develop a new multi-epitope vaccine that could eliminate the possibility of virulence reversal and enhance innate, humoral, and cell-mediated immunity by utilizing a combination of B cell and T cell epitopes. The key benefit of the multi-epitope vaccine is the lack of any live components, which greatly reduces the risk of illness.
Materials and methodsProtein sequence retrieval, antigenicity, and physicochemical characteristicsThe NCBI database (https://www.ncbi.nlm.nih.gov/protein/AYP74770.1?report=fasta) was used to obtain the complete amino acid sequence of the DENV envelope protein (EP) (Accession No: AYP74770) in FASTA format. The functional and structural characteristics of a protein are a reflection of its physicochemical properties. Consequently, the selected DENV (EP) sequence was subjected to the ProtParam program of the ExPASy Proteomics Server (https://web.expasy.org/protparam/)13 for the evaluation of its physicochemical properties. Physicochemical properties including total amino acid length, molecular weight, theoretical PI, negative and positive residues, solubility, instability index, and half-life were analyzed.13 The antigenicity of the DENV (EP) sequence was also evaluated using the vaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html).14 To validate the antigenicity of the EP, a threshold hold value of 0.4 was employed. The server shows DENV (EP) sequences that have antigenic properties, and the scores are greater than 0.5. This DENV (EP) sequence were chosen for further investigation.
Retrieval of binding site of CD8+, CD4+, lineal B- cell from EP sequenceThe selected sequence of the DENV (EP) was submitted to the IEDB server for predicting MHC-I and MHC-II binding site epitopes using the recommended NetMHCPan 4.1 EL (2020.09 (current)) (http://tools.iedb.org/mhci/)15 and Consensus approach (http://tools.iedb.org/mhcii/)16 methods, respectively. The server provided reference sets with maximal population coverage based on HLA allele frequencies. The MHC-I binding site dataset included three alleles (HLA-A02:01, HLA-A02:02, and HLA-A02:06) with a peptide length of 9.17–20 Epitopes with a percentile rank greater than 0.9 were selected. For the MHC-II binding site21,22, a 15-mer length epitope was predicted using the HLA-DRB101:01 allele reference set. Amino acid sequences with CD8+ and CD4+ epitopes were selected for further analysis. The selected DENV (EP) sequence were also subjected BCpreds (http://ailab-projects1.ist.psu.edu:8080/bcpred/predict.html)23 to predict lineal B-cell epitopes. Predicted lineal B-cell epitopes with a length of 20 amino acids and a cut-off score of more than 0.9 were chosen for next steps.
The predicted T-cell and lineal B-cell epitopes were evaluated for their antigenicity using VaxiJenv2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) at a threshold of 0.4. In addition, the protein sequences of these epitopes were analyzed for allergenic and toxic properties using the AllerTOP v. 2.0 (https://www.ddg-pharmfac.net/AllerTOP/)24 and TOXINPRED (http://crdd.osdd.net/raghava/toxinpred/)25 servers, respectively. Furthermore, these epitopes were compared to human protein sequences using the protein BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) to identify homologous properties. Only epitopes with antigenic properties, non-allergenic, non-toxic, and non-homologous were considered for vaccine development.
Adjuvant selection and vaccine constructionHalobacteriumsalinarum (Accession no: CAA28975) were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/protein/CAA28975.1) in FASTA format. The selected Halobacteriumsalinarum was further subjected into the ExPASy Proteomics Server's ProtParam tool (https://web.expasy.org/protparam/) for physicochemical property evaluation, as previously mentioned (Method 2.1). A multi-epitope vaccine was constructed using adjuvant, T-cell (CD8+, CD4+) and lineal B-cell epitopes with appropriate linkers. Prior to the initial EAAAK linkers epitope, Halobacteriumsalinarum was added as an adjuvant to boost the vaccine's immunogenicity. CD8+ epitopes were linked to AAY linkers throughout this process, while CD4+ epitopes were linked to GPGPG linkers. KK linkers were also employed to fuse lineal B-cell epitopes. Furthermore, as previously indicated, our constructed vaccine was subjected to ExPASy Proteomics Server's ProtParam tool (https://web.expasy.org/protparam/) for evaluation of physicochemical properties. To validate the antigenicity of the constructed vaccine was performed using VaxiJenv2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) at threshold 0.4. The MEME/MAST motif approach was performed using AlgPred https://webs.iiitd.edu.in/raghava/algpred/submission.html) to identify allergenicity of the constructed vaccine.
Protein structure perdition, refinement and validation of constructed vaccineTo predict the secondary structure of the constructed vaccine, the PSIPRED 4.0 servers (http://bioinf.cs.ucl.ac.uk/psipred/)26,27 were used. RaptorX web server (http://raptorx.uchicago.edu/ContactMap/)28 was used to predict the 3D structure of the constructed vaccine. This server uses multiple templates and iterative template stitching to select the best template for building the protein. Final 3D structure of constructed vaccine was selected based on the confidence score, and rank.28 The final 3D structure of the vaccine was refined using the Galaxy Refine program (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE).29 This program utilizes a systematic dynamic analysis to repack the side chains, correct any incorrect loops, and smooth out the framework to improve the accuracy and quality of the model.29 To ensure the accuracy and reliability of the improved vaccine structure, we performed validation using two web servers, ProSA and PROCHECK. The ProSA web server (https://prosa.services.came.sbg.ac.at/prosa.php)30 compares the protein structure with known crystallography structures (X-ray and NMR) to provide scores to measure the quality of the structure.30 The PROCHECK web server (https://saves.mbi.ucla.edu/)31 generates a Ramachandran plot, which shows the number of energetically favored, permissible, and disallowed residues in each region, providing information on the stereochemical quality of the structure.31
Conformational B- cell epitope predictionThe presence of conformational (discrete) B-cell epitopes (CBE) is essential in a vaccine, for stimulating antibody production. Therefore, the refined protein structure was checked for the presence of CBE. The ElliPro framework, available on the IEDB servers, was utilized to estimate the CBE.32
Molecular docking and dynamic stimulationMolecular docking is an analytical technique used to determine the binding energy and interaction between a ligand and receptor. Toll-like receptors (TLRs 2 & 7) play a vital role in identifying DENV and activating the monocyte gate. We retrieved the TLR2 (PDB ID: 6NIG) and TLR7 (PDB ID: 6LW1) proteins from the RCSB PDB website. The web-based Pathdock docking server (https://bioinfo3d.cs.tau.ac.il/PatchDock/)33,34 was used to predict the binding affinity and interaction between the constructed vaccine and the TLR2/TLR7 receptors. The best-docked complex with an acceptable pose was derived based on the global energy rating.
To assess the structural integrity and stability of the docked complex, we performed molecular dynamics (MD) simulations. The iMODS server (http://imods.chaconlab.org/)35 used to evaluate deformation and variations in protein mobility by utilizing deformability, Eigenvalues, and the normal mode analysis (NMA) approach.35
Evaluation of immune stimulationOur constructed was submitted to the C-ImmSim server (http://150.146.2.1/C-IMMSIM/)36 to evaluate the vaccine's potential to stimulate an immune response.36 The C-ImmSim server detects epitopes and subsequent immune reactions using machine learning methods and position-specific scoring matrix (PSSM) for agent-based dynamics simulation. Typically, vaccines are administered in three doses separated by 28 days. Therefore, we decided to administer three injections (concentrations) with a four-week break between them. In the C-ImmSim immunological simulator, each time-step corresponds to 8 h in real life. Thus, three injections require 1, 84, and 170 time-steps, respectively. Moreover, we assessed antigen exposure in a typical endemic zone using three injections.
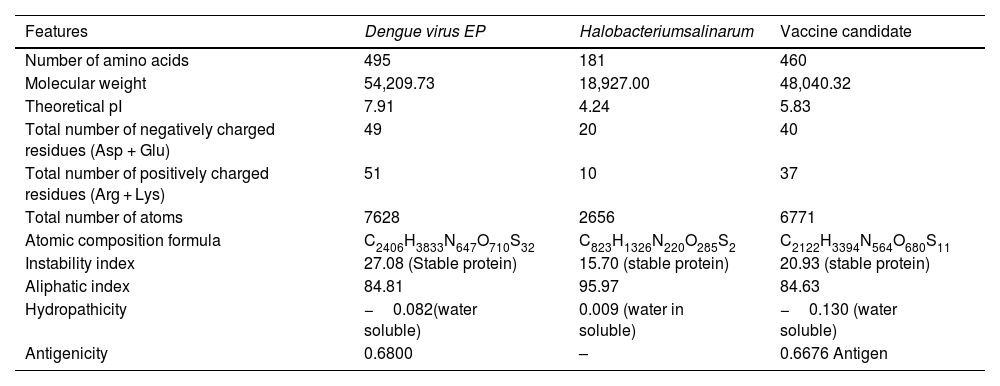
ResultsAnalysis of retrieved protein sequenceThe DENV EP comprises 495 amino acids and has a molecular weight of 54,209.73 kDa, as determined by its physicochemical properties through Protparam analysis. Vaxijen v2.0 analysis confirmed that DENV (EP) is antigenic. The protein has a theoretical isoelectric point (pI) of 7.91, indicating is positively charged. Proteins with isoelectric points above 7 are considered positively charged. Of the total 100 amino acid charges, 51 were found as positive while 49 were negative. The instability score is 27.08, indicating the protein is stable. The aliphatic index is 84.81, indicating a relatively high amount of aliphatic side chains and solubility is −0.082, indicating the protein sequence is water-soluble. The total numbers of Oxygen (O), Carbon (C), Hydrogen (H) Sulfur (S), and Nitrogen (N), were entitled by the formula: C2406H3833N647O710S32 (Table 1).
Evaluation of physicochemical properties of DENV EP, Halobacteriumsalinarum and Vaccine candidate.
| Features | Dengue virus EP | Halobacteriumsalinarum | Vaccine candidate |
|---|---|---|---|
| Number of amino acids | 495 | 181 | 460 |
| Molecular weight | 54,209.73 | 18,927.00 | 48,040.32 |
| Theoretical pI | 7.91 | 4.24 | 5.83 |
| Total number of negatively charged residues (Asp + Glu) | 49 | 20 | 40 |
| Total number of positively charged residues (Arg + Lys) | 51 | 10 | 37 |
| Total number of atoms | 7628 | 2656 | 6771 |
| Atomic composition formula | C2406H3833N647O710S32 | C823H1326N220O285S2 | C2122H3394N564O680S11 |
| Instability index | 27.08 (Stable protein) | 15.70 (stable protein) | 20.93 (stable protein) |
| Aliphatic index | 84.81 | 95.97 | 84.63 |
| Hydropathicity | −0.082(water soluble) | 0.009 (water in soluble) | −0.130 (water soluble) |
| Antigenicity | 0.6800 | – | 0.6676 Antigen |
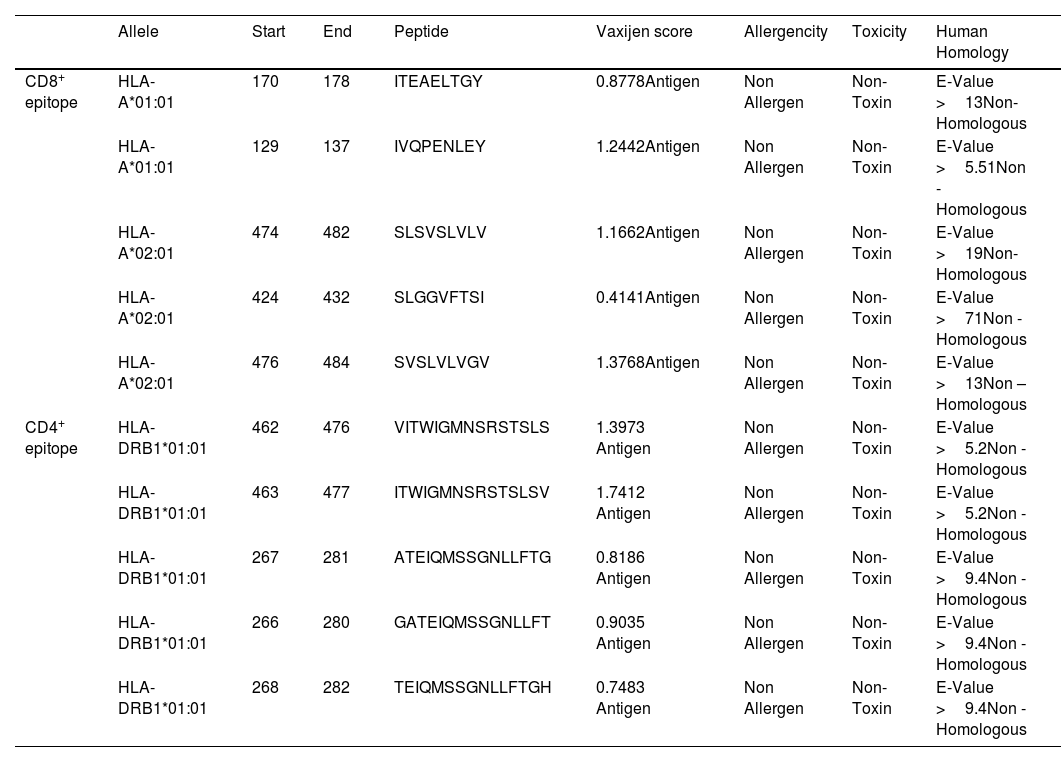
The antigenicity, homology, toxicity, and allergenicity properties of anticipated CD4+, CD8+, and lineal B-cell epitopes were validated using the appropriate server. Antigenic, non-allergic, non-toxic, and non-homologous properties were found in five CD8 + epitopes, five CD4 + epitopes, and five Linear B-cell epitopes (Tables 2 and 3).
Detailed analysis of CD8+& CD4+ epitope.
| Allele | Start | End | Peptide | Vaxijen score | Allergencity | Toxicity | Human Homology | |
|---|---|---|---|---|---|---|---|---|
| CD8+ epitope | HLA-A*01:01 | 170 | 178 | ITEAELTGY | 0.8778Antigen | Non Allergen | Non-Toxin | E-Value >13Non- Homologous |
| HLA-A*01:01 | 129 | 137 | IVQPENLEY | 1.2442Antigen | Non Allergen | Non-Toxin | E-Value >5.51Non -Homologous | |
| HLA-A*02:01 | 474 | 482 | SLSVSLVLV | 1.1662Antigen | Non Allergen | Non-Toxin | E-Value >19Non- Homologous | |
| HLA-A*02:01 | 424 | 432 | SLGGVFTSI | 0.4141Antigen | Non Allergen | Non-Toxin | E-Value >71Non -Homologous | |
| HLA-A*02:01 | 476 | 484 | SVSLVLVGV | 1.3768Antigen | Non Allergen | Non-Toxin | E-Value >13Non –Homologous | |
| CD4+ epitope | HLA-DRB1*01:01 | 462 | 476 | VITWIGMNSRSTSLS | 1.3973 Antigen | Non Allergen | Non-Toxin | E-Value >5.2Non -Homologous |
| HLA-DRB1*01:01 | 463 | 477 | ITWIGMNSRSTSLSV | 1.7412 Antigen | Non Allergen | Non-Toxin | E-Value >5.2Non -Homologous | |
| HLA-DRB1*01:01 | 267 | 281 | ATEIQMSSGNLLFTG | 0.8186 Antigen | Non Allergen | Non-Toxin | E-Value >9.4Non -Homologous | |
| HLA-DRB1*01:01 | 266 | 280 | GATEIQMSSGNLLFT | 0.9035 Antigen | Non Allergen | Non-Toxin | E-Value >9.4Non -Homologous | |
| HLA-DRB1*01:01 | 268 | 282 | TEIQMSSGNLLFTGH | 0.7483 Antigen | Non Allergen | Non-Toxin | E-Value >9.4Non -Homologous |
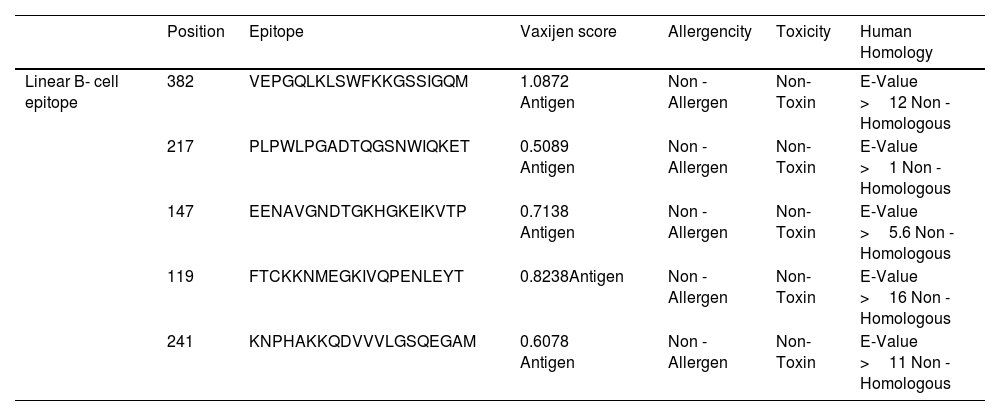
Detailed analysis of Linear B- cell epitope.
| Position | Epitope | Vaxijen score | Allergencity | Toxicity | Human Homology | |
|---|---|---|---|---|---|---|
| Linear B- cell epitope | 382 | VEPGQLKLSWFKKGSSIGQM | 1.0872 Antigen | Non - Allergen | Non-Toxin | E-Value >12 Non -Homologous |
| 217 | PLPWLPGADTQGSNWIQKET | 0.5089 Antigen | Non - Allergen | Non-Toxin | E-Value >1 Non -Homologous | |
| 147 | EENAVGNDTGKHGKEIKVTP | 0.7138 Antigen | Non - Allergen | Non-Toxin | E-Value >5.6 Non -Homologous | |
| 119 | FTCKKNMEGKIVQPENLEYT | 0.8238Antigen | Non - Allergen | Non-Toxin | E-Value >16 Non -Homologous | |
| 241 | KNPHAKKQDVVVLGSQEGAM | 0.6078 Antigen | Non - Allergen | Non-Toxin | E-Value >11 Non -Homologous |
The Methods section 2.3 describes the development of a vaccine using selected CD4+, CD8+, and Linear B cell epitopes along with appropriate linkers. To enhance the adjuvant effect of the vaccine, Halobacteriumsalinarum was added. The physiochemical characteristics of Halobacteriumsalinarum were analyzed using Protparam. The analysis revealed that it has 181 amino acids with a molecular weight of 18,927.00 kDa. The protein has a theoretical isoelectric point (PI) of 4.24, indicating that it is negatively charged. This means that the protein has an isoelectric point <7. Ten amino acid charges were recorded as positive, while 20 amino acid charges were showed as negative. The estimated instability score of Halobacteriumsalinarum is 15.70, indicating a stable protein. The aliphatic-index is 95.97, and the equivalent quantity of aliphatic side chain holding and solubility is 0.009, indicating that the protein sequence is lipid-soluble. The total numbers of Oxygen (O), Carbon (C), Hydrogen (H) Sulfur (S), and Nitrogen (N), were entitled by the formula: C823H1326N220O285S2 (Table 1).
The chosen adjuvant passed all formats of physicochemical properties. The structural and functional studies of the protein suggest that it could be used as an adjuvant. Therefore, Halobacterium salinarum was selected as an adjuvant using the Protparam database. We evaluated the physicochemical parameters of our vaccine and found that it has 460 amino acids with a molecular weight of 48,040.32 kDa. Antigenic properties were evaluated using Vaxijen v2.0, revealed that vaccine shows an antigenic nature. The theoretical isoelectric point (PI) of the developed vaccine was found to be 5.83, indicating its negative charged. If the isoelectric point is lesser than 7, the protein is negatively charged. Positive charges were found nearly 37 amino acids, while 40 were found as negative. Protparam estimated the instability index to be 20.93, indicating that the protein is stable. The aliphatic index of 84.63 suggests that the constructed protein sequence was water-soluble in nature, with a solubility of −0.130. The total numbers of Oxygen (O), Carbon (C), Hydrogen (H) Sulfur (S), and Nitrogen (N), were entitled by the formula: C2122H3394N564O680S11Table 1. As a result of the thorough evaluation, we have determined that our vaccine is a good candidate vaccine (Table 1).
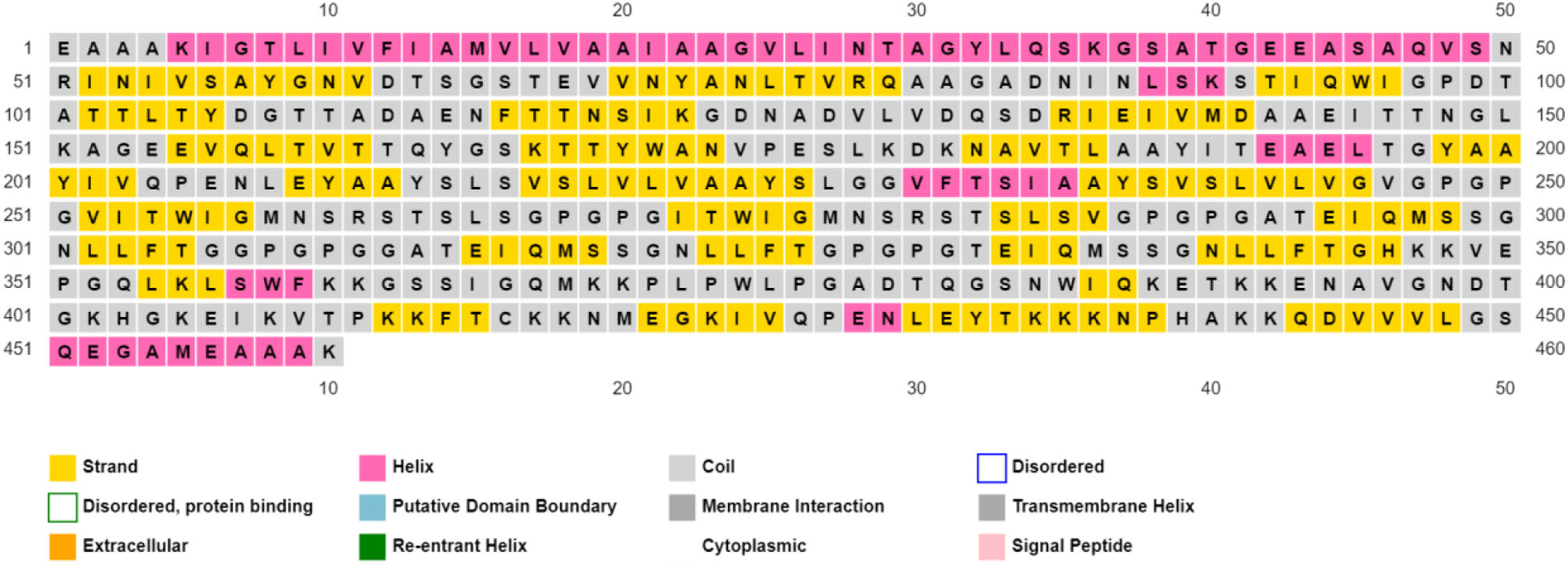
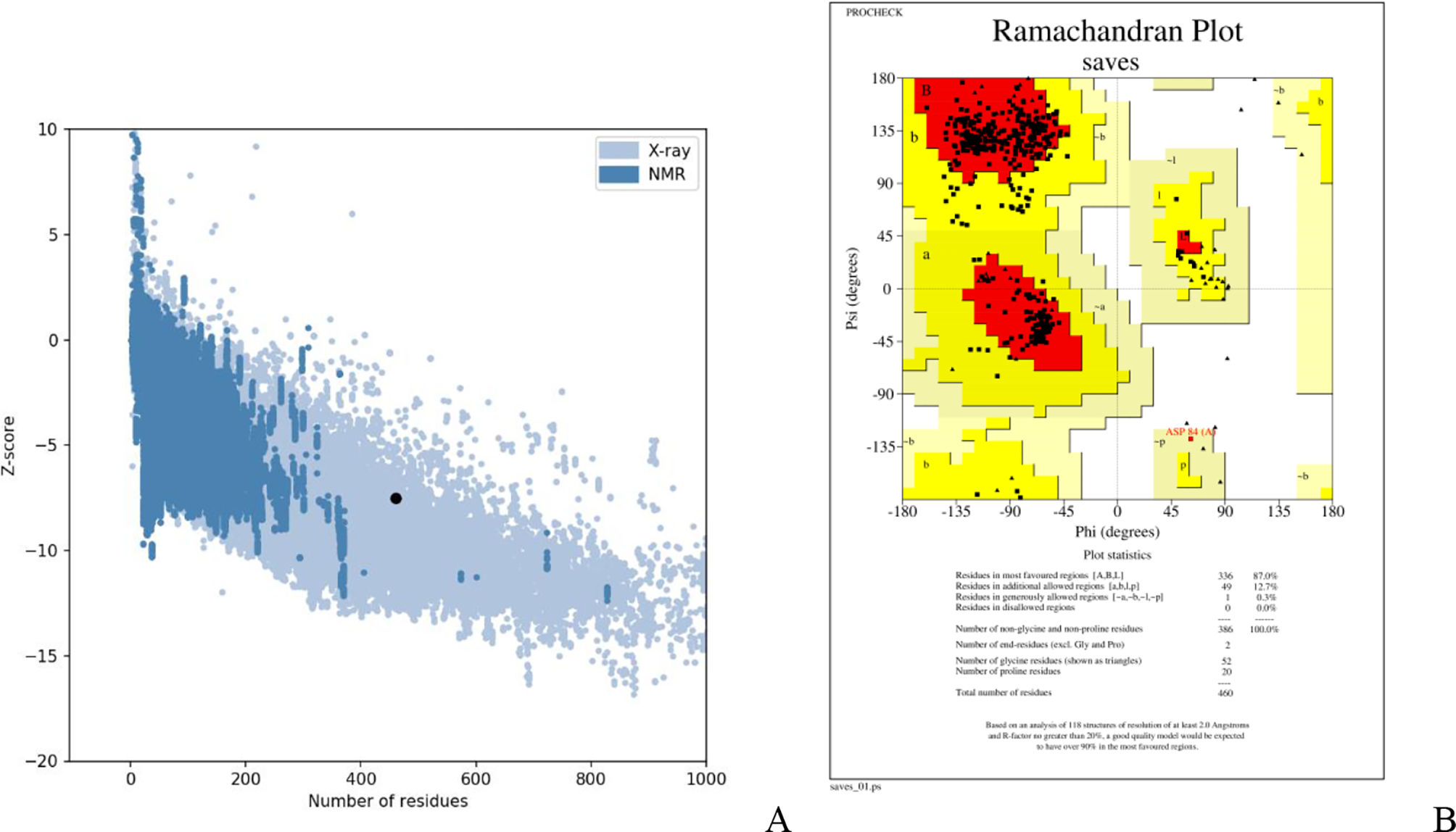
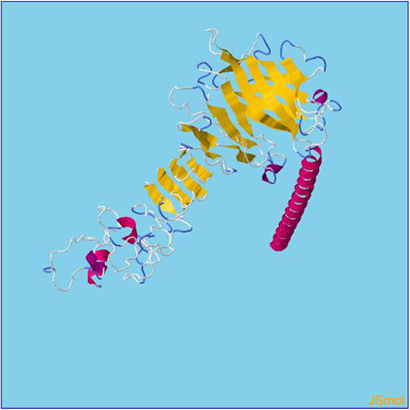
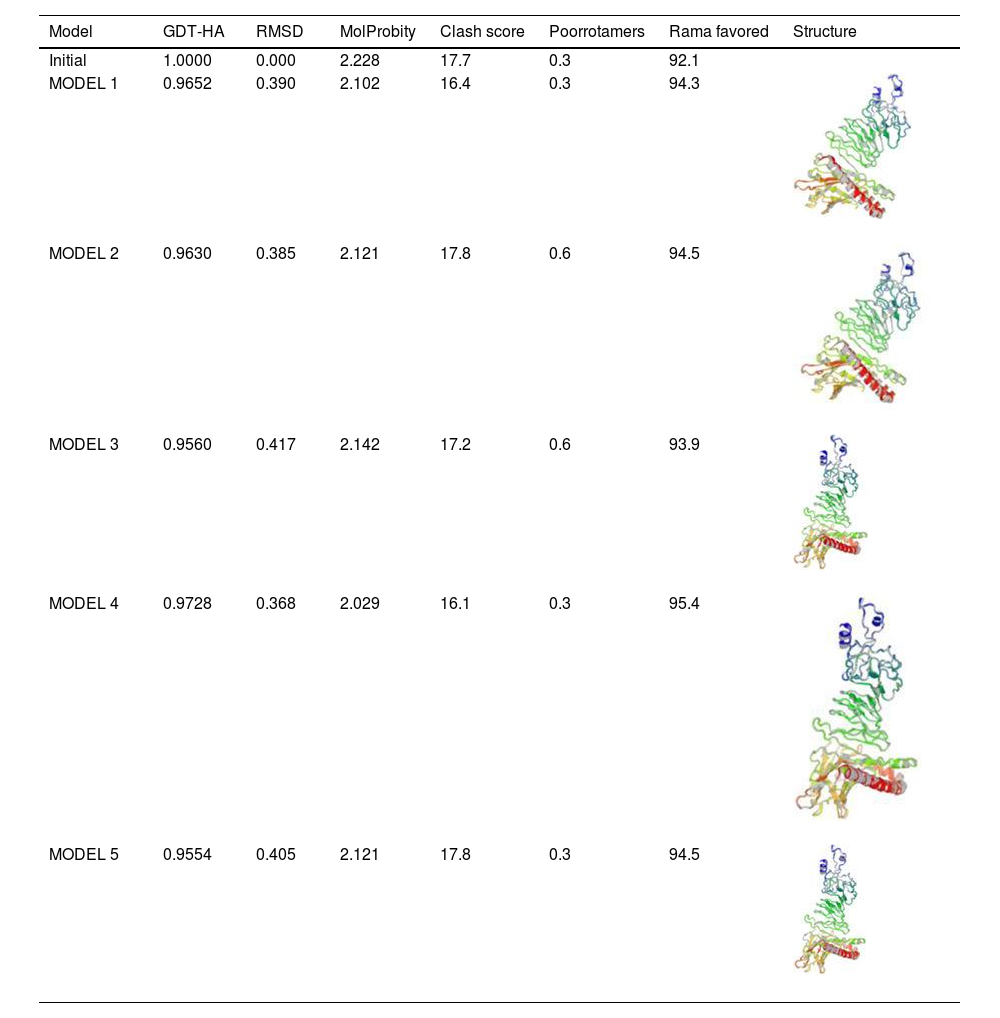
Assessment of protein structure perdition, refinement and validation of constructed vaccinePSIPRED was used to examine the secondary structure of the constructed vaccine (Fig. 1). Alpha helix (23.70%), 310 helix (0.00%), Pi helix (0.00%), Beta Bridge (0.00%), extended strand (28.70%), Beta turns (10.87%), Bend region (0.00%), Random coil (36.74%), Ambiguous states (0.00%). In addition, the constructed vaccine was submitted to the RaportX server for tertiary structure prediction. The RaportX server yielded five tertiary structure models (table 4). We chose the first rank of tertiary structure of the constructed vaccine, which was then refined using the Galaxy web server and this sever generates five refined models. Table 5. The improved model's quality was evaluated in the next section. First, the Global Distance Test (GDT-HA) is used to calculate the correlation between two protein structures using a high precision score. Between five revised models and initial model, the GDT-HA score reaches a maximum of 0.900, indicating that they are quite comparable. The lower the RMSD value, the more stable the system is, and an RMSD score of 0.4 to 0.3 is ideal. The RMSD of the five refined models is less than 0.5, indicating that the protein model is stable. The Molprobity score represents a revised model of crystallographic resolution. The MolProbity score for our five refined protein models is 3, which is significantly lower than the initial model and also indicates critical errors in refined models. The Clash Score represents the amount of unfavorable steric all-atom lie on top, and the refinement improved the model's stability by increasing the clash score from 16 to 17%. The Ramachandran plot score indicates the range of regions that are strongly favored, and a value of more than 85% is usually optimal. However, all revised models pass in all parameters based on their superiority score. As a result, a PDB model of the improved protein structure was obtained for further study Table 3. The ProSA-web server was used to validate the refined final vaccination model consistency, which falls within the score range of −7.5 Fig. 2A. The PROCHEK server was used to perform a Ramachandran plot verification on the refined PDB model. It was found that 87% (336 amino acids) were found in the preferred region, 12.7% (49 amino acids) in the allowed region, and 0.3% (1 amino acid) in the forbidden zone. More than 90% of residues were found to be positioned on acceptable (favorable and permitted) regions in the modified model Fig. 2B.


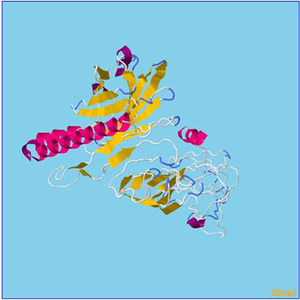
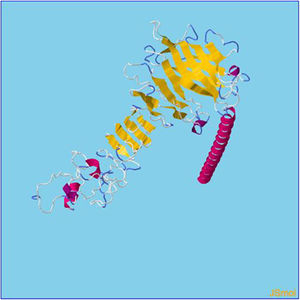
Refinement of constructed vaccine.
| Model | GDT-HA | RMSD | MolProbity | Clash score | Poorrotamers | Rama favored | Structure |
|---|---|---|---|---|---|---|---|
| Initial | 1.0000 | 0.000 | 2.228 | 17.7 | 0.3 | 92.1 | |
| MODEL 1 | 0.9652 | 0.390 | 2.102 | 16.4 | 0.3 | 94.3 | |
| MODEL 2 | 0.9630 | 0.385 | 2.121 | 17.8 | 0.6 | 94.5 | |
| MODEL 3 | 0.9560 | 0.417 | 2.142 | 17.2 | 0.6 | 93.9 | |
| MODEL 4 | 0.9728 | 0.368 | 2.029 | 16.1 | 0.3 | 95.4 | |
| MODEL 5 | 0.9554 | 0.405 | 2.121 | 17.8 | 0.3 | 94.5 |

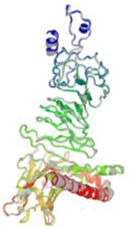
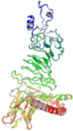

β- Strand structure is denoted as yellow color amino acids, α – helix structure is denoted as pink color amino acids and coil structure is denoted as ash color amino acids.
There are five models of 3D protein structure was predicted and it also categorized by the Rank and estimated RMSD(Å) value. Moreover, rank 1 shows the estimated RMSD(Å) value was found to be 9.1928followed by Rank 2 shows the estimated RMSD(Å) value was found to be 10.378 followed by Rank 3 shows the estimated RMSD(Å) value was found to be 11.434 followed by Rank 4 shows the estimated RMSD(Å) value was found to be 10.726. Finally, Rank 5 shows the estimated RMSD(Å) value was found to be 9.9825.
Table 5. Refinement of constructed vaccine. There are five models of refined 3D protein structure was predicted and it also categorized by the model,GDT-HA, RMSDvalue, MolProbity, Clashscore, Poor rotamers. Moreover, model 1 shows the shows the GDT-HA (0.9652), RMSDvalue (0.390),MolProbity (2.102),Clash score (16.4),Poor rotamers (0.3), Ramachandran plot (94.3) followed by model 2 shows the GDT-HA (0.9630), RMSDvalue (0.385), MolProbity (2.121),Clash score (17.8),Poor rotamers (0.6), Ramachandran plot (94.5)followed by Model 3 shows the GDT-HA (0.9560), RMSDvalue (0.417), MolProbity (2.142),Clash score (17.2),Poor rotamers (0.6), Ramachandran plot (93.9)followed by model 4 shows the GDT-HA (0.9728), RMSDvalue (0.368),MolProbity (2.029),Clash score (16.1),Poor rotamers (0.3), Ramachandran plot (95.4). Finally model 5 shows the GDT-HA (0.9554), RMSDvalue (0.405),MolProbity (2.121),Clash score (17.8) Poor rotamers (0.3), Ramachandranplot (94.5).
The Z-score of the refined model that falls within the score range is −7.5.Ramachandran plot study indicates 87%, 12.7%,and 0.3% of protein residues in favored, allowed and disallowed (foreign) regions, individually.
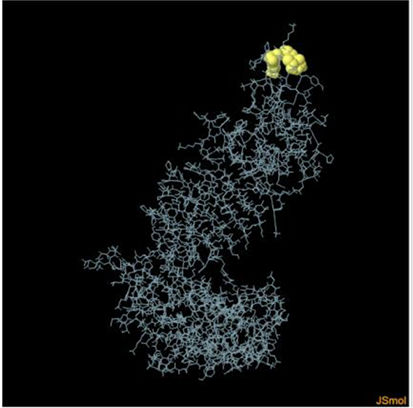
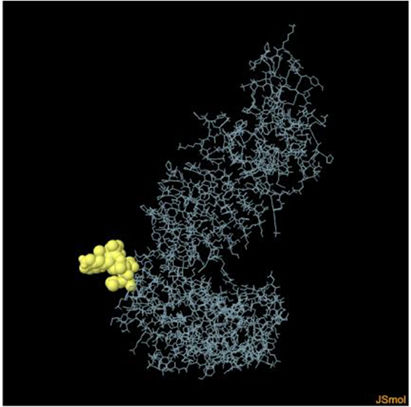
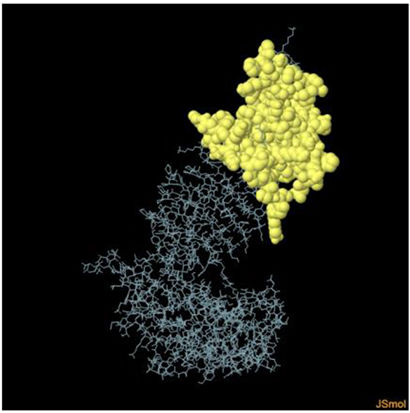
Assessment of conformational B-cell epitopeRefined vaccine model subjected to ElliPro server to identify CBC epitopes. The ElliPro server predicted five new CBC epitopes involving 266 residues with scores ranging from 0.6 to 0.9. Table 6 shows the whole 3D model as well as the details for those four epitopes.
It represents conformation of linear B- cell epitope.
| No | Residues | Number of Residues | Score | 3D Structure |
|---|---|---|---|---|
| I | A:T433, A:K434, A:K436 | 3 | 0.988 | |
| II | A:E1, A:A2, A:A3, A:A4, A:K5, A:I6, A:G7, A:T8, A:L9 | 9 | 0.826 | |
| III | A:V349, A:E350, A:P351, A:G352, A:Q353, A:K355, A:K361, A:G362, A:S363, A:S364, A:I365, A:G366, A:Q367, A:K369, A:K370, A:P371, A:L372, A:P373, A:W374, A:L375, A:P376, A:G377, A:Q381, A:S383, A:I386, A:Q387, A:K388, A:T390, A:K391, A:K392, A:E393, A:N394, A:A395, A:V396, A:G397, A:N398, A:D399, A:T400, A:G401, A:K402, A:H403, A:G404, A:K405, A:E406, A:I407, A:K408, A:V409, A:T410, A:P411, A:K412, A:K413, A:F414, A:T415, A:C416, A:K417, A:N419, A:M420, A:G422, A:K423, A:I424, A:V425, A:Q426, A:P427, A:E428, A:N429, A:L430, A:E431, A:Y432, A:N437, A:P438, A:H439, A:A440, A:K441, A:K442, A:Q443, A:D444, A:V445, A:V446, A:V447, A:L448, A:G449, A:S450, A:Q451, A:E452, A:G453, A:A454, A:M455, A:E456, A:A457, A:A458, A:A459, A:K460 | 92 | 0.693 | |
| IV | A:F12, A:I13, A:V16, A:A19, A:A20, A:A22, A:A23, A:G24, A:L26, A:I27, A:N28, A:T29, A:A30, A:G31, A:Y32, A:L33, A:Q34, A:S35, A:K36, A:G37, A:S38, A:A39, A:T40, A:G41, A:E42, A:E43, A:A44, A:S45, A:A46, A:Q47, A:S49, A:N50, A:R51, A:N60, A:V61, A:D62, A:T63, A:S64, A:G65, A:S66, A:T67, A:E68, A:V69, A:N71, A:Y72, A:A80, A:A81, A:G82, A:A83, A:D84, A:N85, A:I86, A:N87, A:L88, A:S89, A:K90, A:S91, A:T92, A:G97, A:P98, A:D99, A:T100, A:A101, A:T102, A:T103, A:L104, A:T105, A:Y106, A:D107, A:G108, A:T109, A:T110, A:A111, A:D112, A:A113, A:E114, A:N115, A:F116, A:T117, A:T118, A:N119, A:S120, A:I121, A:K122, A:G123, A:D124, A:N125, A:A126, A:D127, A:V128, A:L129, A:V130, A:D131, A:Q132, A:S133, A:D134, A:V139, A:D141, A:A142, A:A143, A:E144, A:I145, A:T146, A:T147, A:N148, A:G149, A:L150, A:K151, A:A152, A:G153, A:E154, A:T161, A:T162, A:Q163, A:Y164, A:G165, A:S166, A:V174, A:P175, A:E176, A:K179, A:D180, A:K181, A:N182, A:A183, A:A193, A:E194, A:L195, A:Q204, A:P205, A:N207, A:L208, A:E209, A:Y210, A:A212, A:Y213, A:V222, A:A223, A:A224, A:Y225, A:S226, A:L227, A:G228, A:G229, A:V230, A:F231, A:T232, A:S233, A:I234, A:A235, A:A236, A:Y237, A:V246, A:G247, A:P248, A:G249, A:P250, A:P268, A:G269, A:P270, A:G271, A:I272 | 162 | 0.661 |
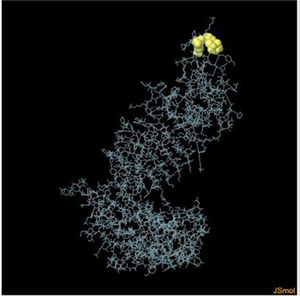
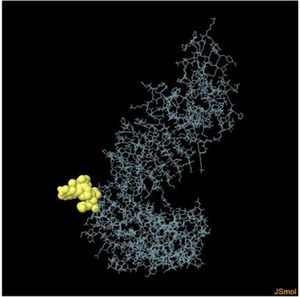
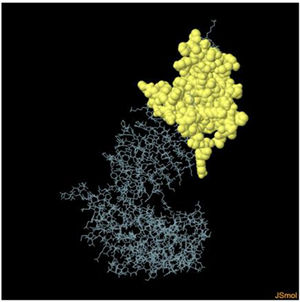
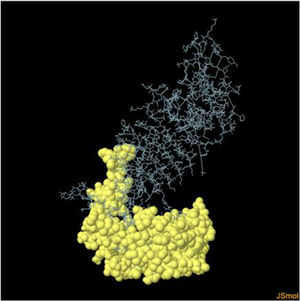
Table 6. It represents conformation of lineal B- cell epitope. The 3D model of the 5 predicted CBC epitopes in the refined vaccine model. The yellow parts are representing the CBC epitopes and the gray parts are represents the other residues. (I) 3 residues of CBC fall within a score of 0.988. (II) 9 residues of CBC fall within a score of 0.826. (III) 92 residues of CBC fall within a score of 0.693. (IV) 162 residues of CBC fall within a score of 0.661.
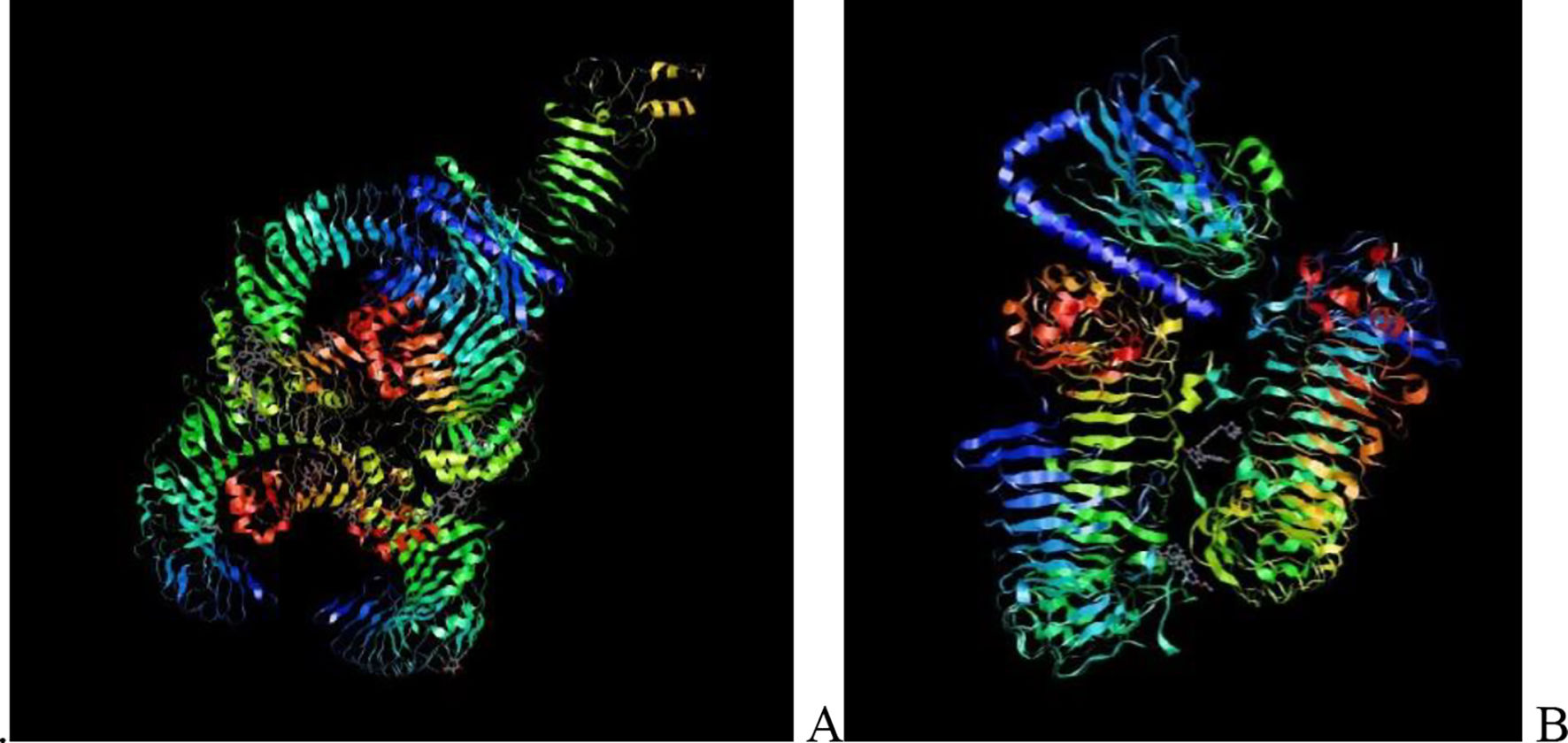
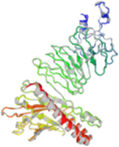
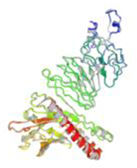
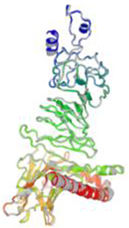
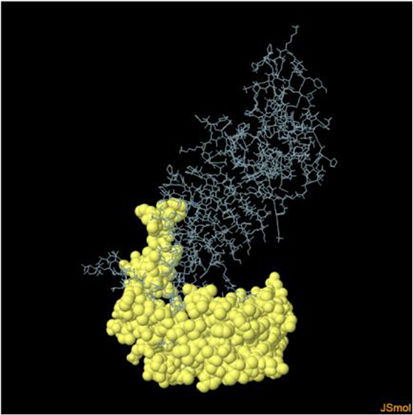
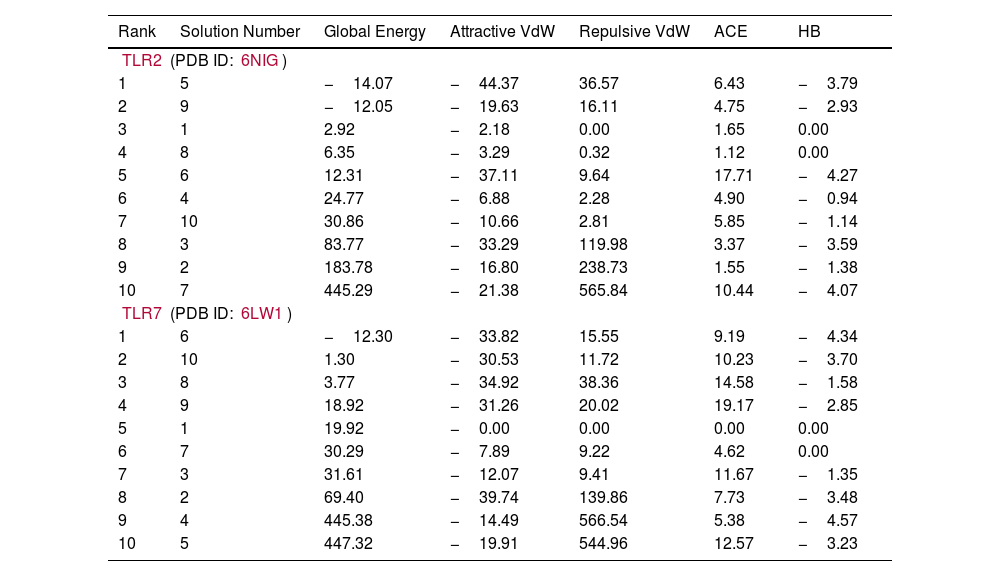
Molecular docking and dynamic stimulationMolecular docking was used to evaluate the stability and binding interaction of the docked complex. TLR2, TLR7, and other important human proteins were employed to identify the immunological response against DENV. TLR2 and TLR7 were chosen as the immunological receptors for molecular docking. The Pathdock server was used to execute molecular docking between the revised 3D model of our final vaccine and the immunological receptors TLR2 (PDB ID: 6NIG) and TLR7 (PDB ID: 6LVX). From the whole docking model, we chose the lowest energy score of −7.87 (Fig. 3A), −43.57 (Fig. 3B), and (Table 7) as the best docked complex, indicating strong binding affinity. Using the iMOD server and MD simulation, we assessed the stability and physical motions of the vaccine-TLR2/TLR7 (docked complex). However,TLR2 showed stronger stability and physical movements compared to TLR7 (Fig. 4).
")
The docking complex of theTLR2, TLR7 and the vaccine model. The vaccine protein indicates combination of red-gray color peptide and the TLR2, TLR7 receptor indicates remaining residue. This complex model's lowest energy score is –7.87,–43.57 global energy suggesting strong binding affinity. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It represents binding affinity between our construted vaccine and TLR2, TLR7.
| Rank | Solution Number | Global Energy | Attractive VdW | Repulsive VdW | ACE | HB |
|---|---|---|---|---|---|---|
| TLR2 (PDB ID: 6NIG) | ||||||
| 1 | 5 | −14.07 | −44.37 | 36.57 | 6.43 | −3.79 |
| 2 | 9 | −12.05 | −19.63 | 16.11 | 4.75 | −2.93 |
| 3 | 1 | 2.92 | −2.18 | 0.00 | 1.65 | 0.00 |
| 4 | 8 | 6.35 | −3.29 | 0.32 | 1.12 | 0.00 |
| 5 | 6 | 12.31 | −37.11 | 9.64 | 17.71 | −4.27 |
| 6 | 4 | 24.77 | −6.88 | 2.28 | 4.90 | −0.94 |
| 7 | 10 | 30.86 | −10.66 | 2.81 | 5.85 | −1.14 |
| 8 | 3 | 83.77 | −33.29 | 119.98 | 3.37 | −3.59 |
| 9 | 2 | 183.78 | −16.80 | 238.73 | 1.55 | −1.38 |
| 10 | 7 | 445.29 | −21.38 | 565.84 | 10.44 | −4.07 |
| TLR7 (PDB ID: 6LW1) | ||||||
| 1 | 6 | −12.30 | −33.82 | 15.55 | 9.19 | −4.34 |
| 2 | 10 | 1.30 | −30.53 | 11.72 | 10.23 | −3.70 |
| 3 | 8 | 3.77 | −34.92 | 38.36 | 14.58 | −1.58 |
| 4 | 9 | 18.92 | −31.26 | 20.02 | 19.17 | −2.85 |
| 5 | 1 | 19.92 | −0.00 | 0.00 | 0.00 | 0.00 |
| 6 | 7 | 30.29 | −7.89 | 9.22 | 4.62 | 0.00 |
| 7 | 3 | 31.61 | −12.07 | 9.41 | 11.67 | −1.35 |
| 8 | 2 | 69.40 | −39.74 | 139.86 | 7.73 | −3.48 |
| 9 | 4 | 445.38 | −14.49 | 566.54 | 5.38 | −4.57 |
| 10 | 5 | 447.32 | −19.91 | 544.96 | 12.57 | −3.23 |
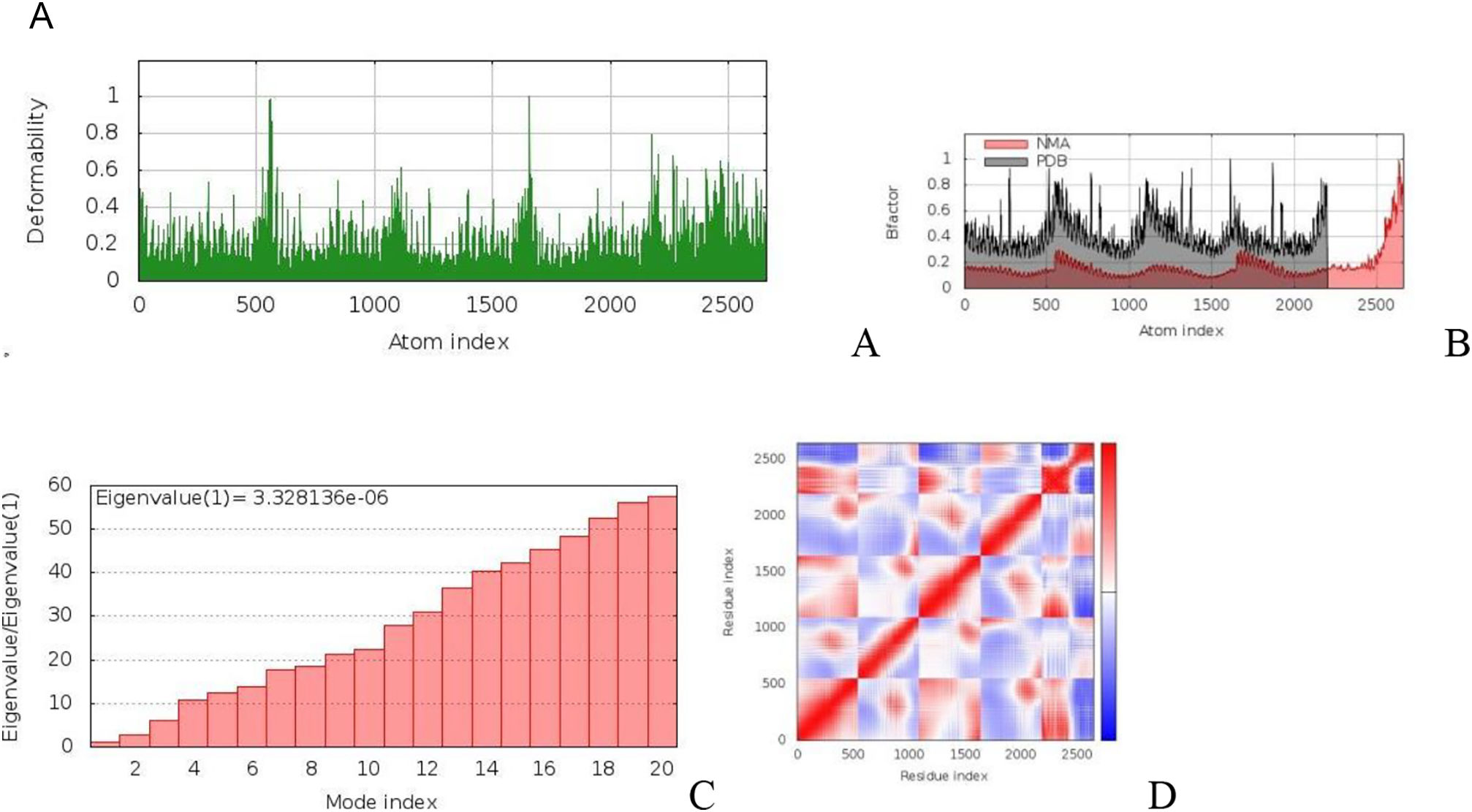
 The main-chain deformability simulation showed the hinges regions are average deformability. (B) B-factor values, determined by normal mode analysis, measure the volatility of each atom. (C) The eigenvalue of the docked complexes demonstrated that does not required energy to deform the structure. (D) Covariance matrix between residue pairs (white: uncorrelated, red: correlated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) Fig. 4b MD modeling of docked vaccine-TLR7 complex complexes. (A) The hinges regions demonstrated strong deformability in the main-chain deformability simulation. (B) B-factor values are used to determine the volatility of each atom using normal mode analysis. (C) The energy required to alter the structure was shown by the eigenvalue of the docked complexes. (D) Matrix of covariance between residue pairs (red: correlated, white: uncorrelated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)")
 The main-chain deformability simulation showed the hinges regions are average deformability. (B) B-factor values, determined by normal mode analysis, measure the volatility of each atom. (C) The eigenvalue of the docked complexes demonstrated that does not required energy to deform the structure. (D) Covariance matrix between residue pairs (white: uncorrelated, red: correlated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) Fig. 4b MD modeling of docked vaccine-TLR7 complex complexes. (A) The hinges regions demonstrated strong deformability in the main-chain deformability simulation. (B) B-factor values are used to determine the volatility of each atom using normal mode analysis. (C) The energy required to alter the structure was shown by the eigenvalue of the docked complexes. (D) Matrix of covariance between residue pairs (red: correlated, white: uncorrelated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)")
a. The simulation of MD of vaccine-TLR2 docked complex structures. (A) The main-chain deformability simulation showed the hinges regions are average deformability. (B) B-factor values, determined by normal mode analysis, measure the volatility of each atom. (C) The eigenvalue of the docked complexes demonstrated that does not required energy to deform the structure. (D) Covariance matrix between residue pairs (white: uncorrelated, red: correlated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) Fig. 4b MD modeling of docked vaccine-TLR7 complex complexes. (A) The hinges regions demonstrated strong deformability in the main-chain deformability simulation. (B) B-factor values are used to determine the volatility of each atom using normal mode analysis. (C) The energy required to alter the structure was shown by the eigenvalue of the docked complexes. (D) Matrix of covariance between residue pairs (red: correlated, white: uncorrelated, blue: anti-correlated). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 7. This table shows the binding affinity between the constructed vaccine and TLR2, TLR7. Global energy was used to classify the binding affinity, with lower global energy indicating higher binding affinity. The Rank 1 PDB format was retrieved for molecular dynamics analysis, with TLR2 Rank 1 having a global energy of −7.87 and TLR7 Rank 1 having a global energy of −43.57.
Fig. 3 shows the docking complex between the vaccine model (represented by a red-gray color peptide) and the TLR2 (A), TLR7(B) receptors (represented by remaining residues). The complex has a low global energy score of −7.87 (A) and −43.57 (B), indicating a strong binding affinity.
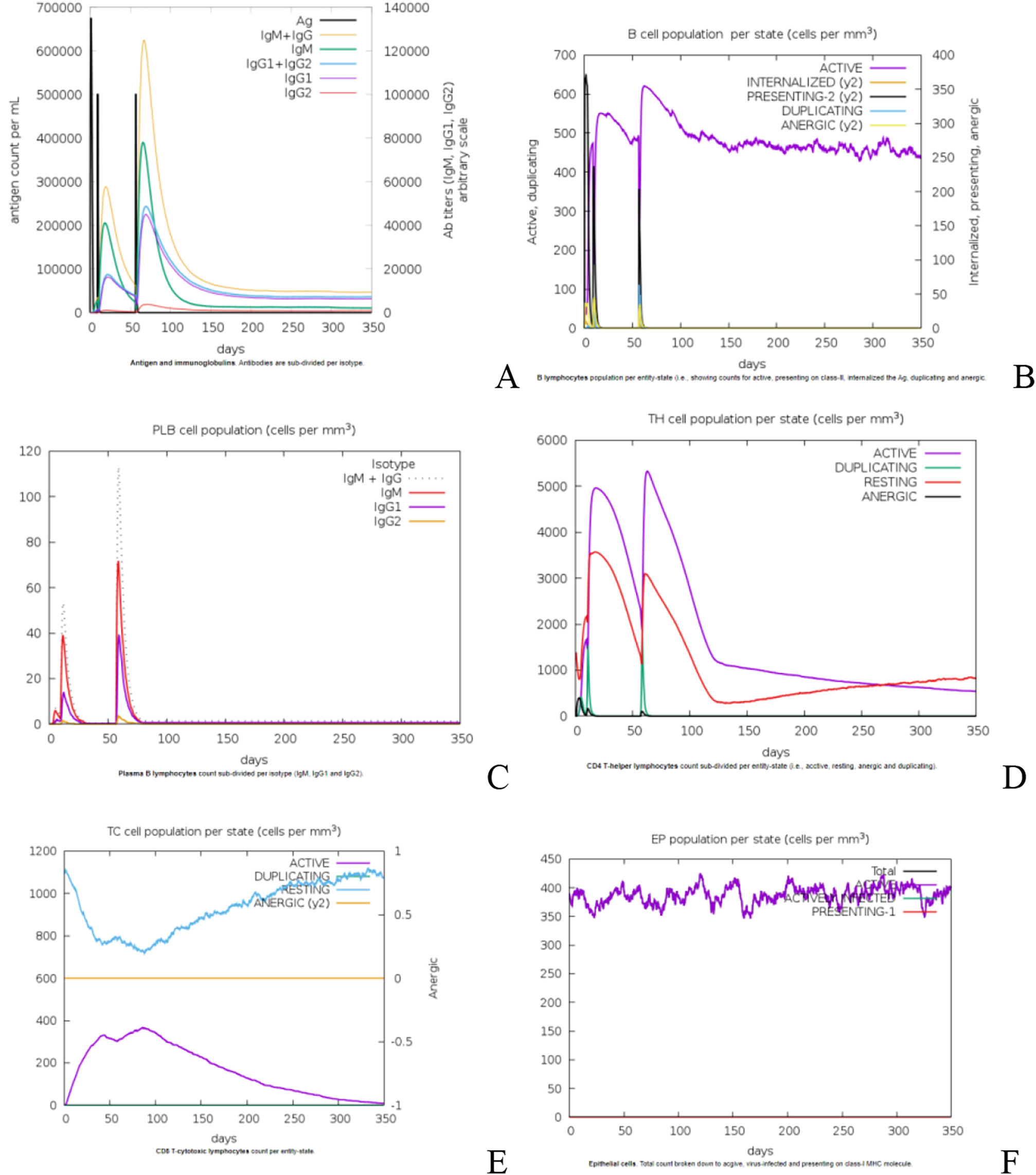
Analysis of immune stimulationTo simulate the immunological response induced by vaccination, three doses of the proposed vaccine were administered four weeks apart at a dose rate of 1050 units per dose. In silico immune simulation plots in a population with a VL susceptible HLA profile after hypothetical immunization, antigenic recognition and subsequent response in terms of antibody production and active as well as memory B cell and T cell generation were indicated. A marked increase in vaccine-specific IgM production can characterize the primary response to the proposed vaccine. IgM + IgG antibodies are rapidly increased in secondary and tertiary responses, followed by IgM and IgG1 + IgG2 antibodies. Fig. 5A shows a comparable drop in antigen concentration following consecutive doses. Fig. 5B depicts the constancy of the effector B-cell during the course of 350 days. The plasma B- cells increase their antibodies rapidly during secondary and tertiary dosage responses, including IgM + IgG antibodies, as illustrated in Fig. 5C. As a result, we concluded that the effector B-cell and Plasma B-cells indicate a consistent increase in the production of persistent memory B cells. Following the initial dose, CD4+ T and CD8+ lymphocyte growth with memory formation was seen, as illustrated in Fig. (5D &E). Interferon gamma and interlukin-2 have the highest levels in the blood plasma after three doses of the vaccination, followed by interleukin −6, 23. In addition, when compared to interleukin-10, interleukin-12 has a lower immunogenic effect, as seen in Fig. 5E.
 Immune response patterns after antigen exposure. (B) The consistency of the effector B-cells (C) Plasma B-cell immune response. (D) The population of cytotoxic CD4+ cells. (E) The population of cytotoxic CD8+ cells. (F) Generation of cytokines.")
A designed vaccination causes immune simulation. (A) Immune response patterns after antigen exposure. (B) The consistency of the effector B-cells (C) Plasma B-cell immune response. (D) The population of cytotoxic CD4+ cells. (E) The population of cytotoxic CD8+ cells. (F) Generation of cytokines.
The DEN-2 sequence was obtained from NCBI to retrieve the envelope protein sequences. T- Cell and B-cell epitopes were predicted using the retrieved protein sequences. CD4+ and CD8+, lineal B cell epitopes were chosen.37,38 To enhances the vaccine's immunogenicity, a short stretch of -defensin amino acid sequence was added to the N-terminal end of the final construct, which will serve as an adjuvant. The construct was also assessed for allergenicity using AllerTOP v. 2.0 and for antigenicity using VaxiJen v2.0. In addition, various physicochemical properties including molecular weight, half-life, sequence length, aliphatic index, instability index, theoretical pI, and grand average of hydropathicity were analyzed using the ExPASy–ProtParam tool. PsiPred was employed to predict the secondary structure of the vaccine construct, while homology modeling was used to generate a simulation of the tertiary structure. RAMPAGE was utilized to assess the predicted tertiary structure for allowed and disallowed regions in the Ramachandran plot.39 After generating the tertiary structure of the vaccine, molecular docking of the vaccine structure with TLR-2 and TLR-7 was performed to examine the interactions between the ligand and receptor molecules. The docked complex was further analyzed using molecular dynamics simulation to validate the stable interaction between the ligand and receptor at the microscopic level.
We specifically chose to target dengue serotype 2 because the existing vaccine, Dengvaxia, was unable to prevent this particular serotype. Our vaccine was designed to be effective against dengue serotype 2 compared to serotypes 1, 3, and 4. This is similar to how the COVID-19 vaccine was unable to protect against the double mutant strain of the virus. Although the replication process is the same for all dengue serotypes, the morphology of the virus, particularly the outer layer of the viral protein, differs between each serotype as a mechanism to protect itself from immune cells, known as the virus escape mechanism.40 Therefore, we selected the outer layer of the viral protein that differs between each serotype, making our constructed vaccine specifically effective against dengue serotype 2.
As we know, live attenuated or non-attenuated vaccines have some drawbacks as they contain genetic material of specific viruses, which may become active when injected into the human body and cause harmful effects. However, multi-epitope vaccines only contain the outer layer of the viral protein, where the immune system can bind and activate against the specific virus. This is why we specifically focused on developing a multi-epitope vaccine. In silico development of a multi-epitope vaccine candidate is more important in therapeutics than single epitope development because it makes it easier to find antigenic epitopes that could trigger a tailored immune response.
Furthermore, the immune-informatics strategy for designing such a candidate ensures a cost-effective and time-saving vaccine preparation process. Since the design comprises both T and B epitopes, a multi-epitope vaccine construct was developed that could potentially trigger both humoral and cell-mediated immune responses upon injection into the host. In terms of targeted immunological features, the proposed vaccine is likely to provide double protection. The vaccine candidate includes epitopes for numerous viral proteins, which could provide significant protection against infection. The construct was developed using envelope proteins with conserved sequences specific to dengue serotype 2. The vaccine was composed of T-cell and B-cell epitopes and were linked together by linkers.
ConclusionIn conclusion, many vaccine inventions fail in clinical trials due to issues such as allergic, toxic, non-antigenic properties, and poor physicochemical properties. One reason for this is the use of an unstable strain, which can make it difficult to isolate a particular antigenic protein. However, to overcome these problems, Insilico studies play a significant role. Our constructed vaccine demonstrates favorable physicochemical properties, is non-allergic, non-toxic, non-homologous, and specifically designed for the dengue virus 2 strain. In other words, Insilico methods can significantly increase the success probability for new vaccine inventions, saving time and resources. In the future, these research findings will be useful in taking this research to the next level with in vitro and in vivo studies.
CRediT authorship contribution statementMohamed Sheik Tharik Abdul Azeeze: Writing - original draft, Methodology, Formal Analysis, Validation and Supervision.
Rajaguru Arivuselvam: Interpretation, writing and supervision.
Consent for publicationNot applicable.
FundingNone.
Ethical approvalNot required.
Authors would like to thank JSSAHER, Mysuru for the facilities offered.