




La neoplasia hematodérmica CD4/CD56+ o de células dendríticas plasmocitoides blástica es un tumor agresivo, muy infrecuente, derivado de precursores de células dendríticas plasmocitoides1. Conlleva muy mal pronóstico sin tratamiento, y la mediana de supervivencia es de 12 a 14 meses con quimioterapia intensiva, aunque se han descrito supervivencias prolongadas tras el trasplante alogénico de progenitores hematopoyéticos2. Presentamos los casos de 2 varones que consultaron por lesiones cutáneas asintomáticas, asociadas a citopenias en sangre periférica.
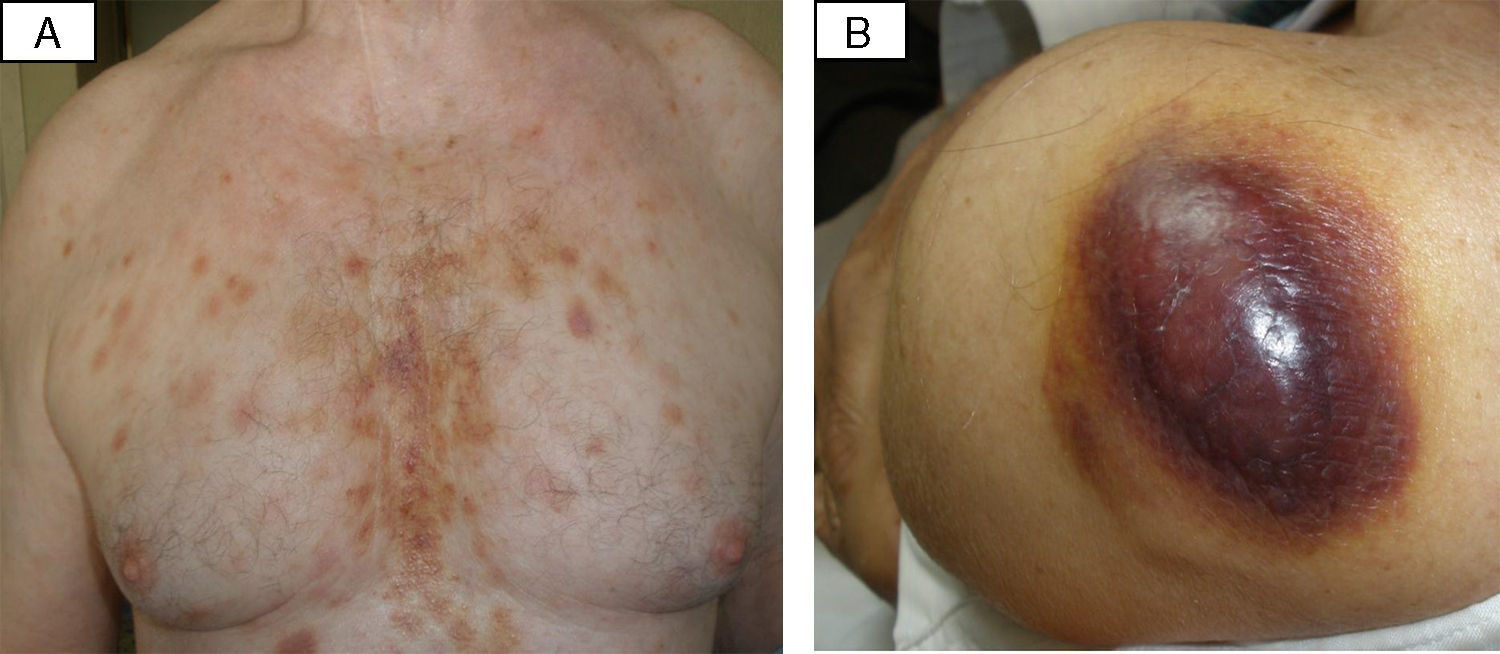
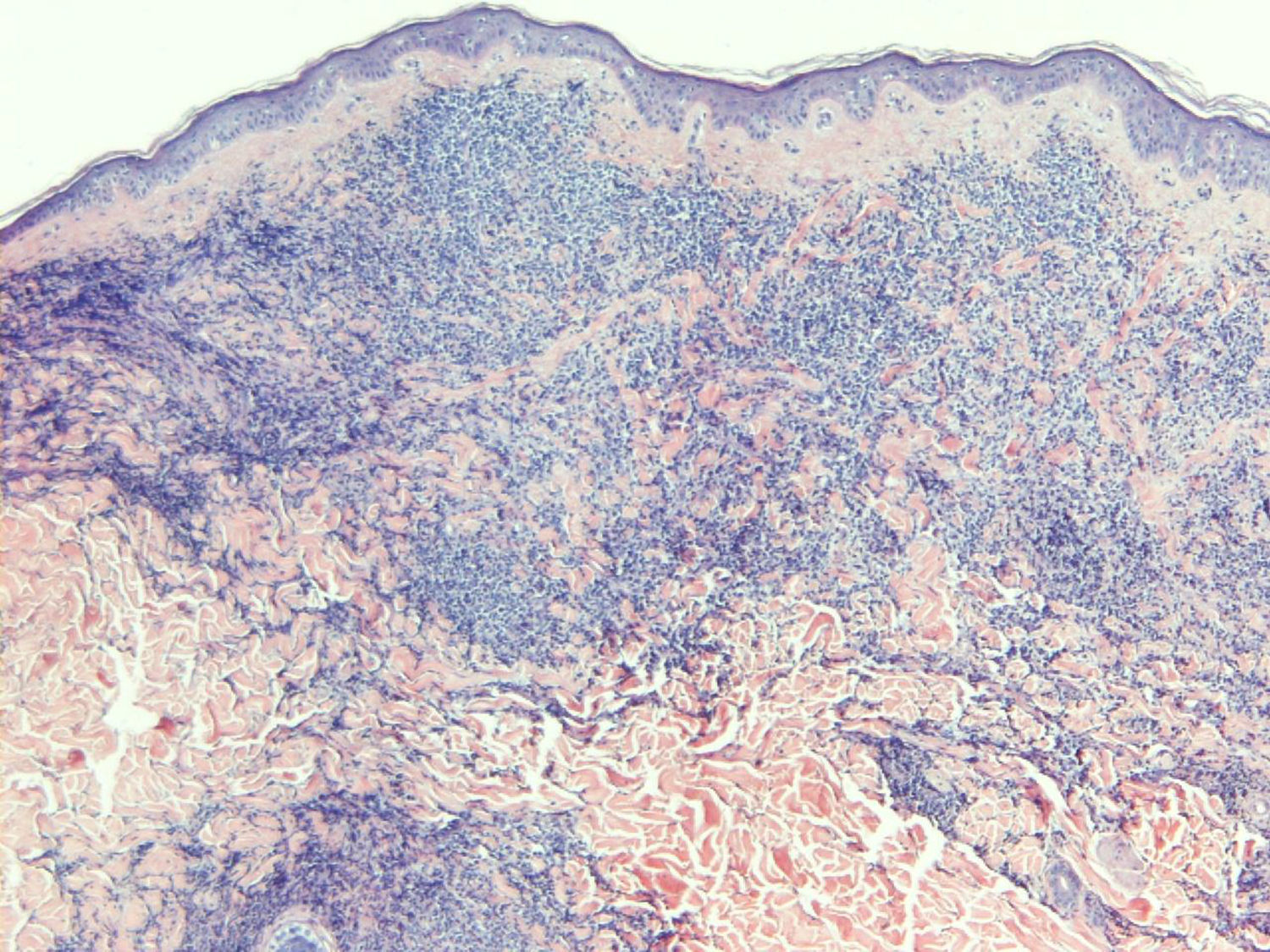
Casos clínicosEl primer paciente es un varón 78 años de edad con antecedentes personales de una anemia refractaria con exceso de blastos de 4 años de evolución, en tratamiento con eritropoyetina y transfusiones periódicas. Consultaba por una lesión cutánea en el hombro izquierdo de un mes de evolución no asociada a un traumatismo previo, coincidente con una anemización importante y pérdida de la respuesta a la eritropoyetina en el contexto de una pancitopenia (7,6g/dl de hemoglobina, 16.000 plaquetas/μl, 1.500 leucocitos/μl). En la exploración física se observó un nódulo infiltrado de tonalidad parduzca (fig. 1). Realizamos una biopsia cutánea que puso de manifiesto la presencia de un infiltrado denso dérmico de células de talla media y citoplasma mal definido (fig. 2). Las técnicas de inmunohistoquímica revelaron que las células se teñían positivamente con los marcadores para CD4, CD43, Bcl2, CD56 y CD123, y negativamente para CD3, CD5, CD7, CD8, CD20 lo que permitió el diagnóstico de infiltración pandérmica por una neoplasia de células dendríticas plasmocitoides blásticas. La biopsia de médula ósea demostró que los megacariocitos estaban muy disminuidos, las series mieloide y eritroide casi ausentes, y la infiltración de un 79% de la misma por células con el mismo inmunofenotipo que en la biopsia cutánea, con una morfología característicamente descrita que recuerda a un espejo de mano. El paciente no presentaba afectación de otros órganos. Dada la edad y comorbilidades del paciente se decidió instaurar únicamente un tratamiento de soporte. El paciente falleció por una neumonía retrocardíaca un mes tras el diagnóstico.

.")
El segundo caso corresponde a un varón de 68 años, sin antecedentes personales de interés, que consultó por lesiones cutáneas asintomáticas de más de un año de evolución. En la exploración se observaron múltiples pápulas y nódulos infiltrados eritematovioláceos y parduzcos, de predominio en el tronco y en la raíz de las extremidades superiores (fig. 1). En la analítica sanguínea solo destacaba la presencia de una discreta anemia (hemoglobina 11,2g/dl) y leucopenia (2.800 leucocitos/μl). La tomografía axial computarizada reveló solo escasos ganglios de tamaño no patológico en las regiones cervical, axilares y mediastínica. Realizamos una biopsia cutánea que mostraba una infiltración de la dermis por células redondeadas de citoplasma escaso y basófilo, con una evidente zona Grenz. Con las técnicas de inmunohistoquímica las células neoplásicas fueron positivas para CD4, CD56, CD68 y Bcl2, siendo negativas para CD3, CD5, CD7, CD8, CD20, CD79a, CD34, P53, TdT, CD99, Bcl6. La tasa de proliferación celular era elevada (>60%). En el aspirado de médula ósea se apreciaba la existencia de un 44% de blastos, compatible con una leucemia aguda con escasa expresión periférica y una citometría acorde.
El paciente fue trasladado al Servicio de Hematología, donde se inició el tratamiento con poliquimioterapia, consiguiéndose una remisión completa morfológica tras dicha quimioterapia de inducción, con una citometría posterior negativa para células dendríticas y una mejoría de las lesiones cutáneas. El paciente recibió posteriormente tratamiento de intensificación y alotrasplante de progenitores hematopoyéticos de donante no emparentado, con supervivencia libre de enfermedad hasta el momento actual (2 años y 5 meses desde el diagnóstico).
ComentarioLa neoplasia de células dendríticas plasmocitoide blásticas es un tumor agresivo derivado de los precursores de las células dendríticas plasmocitoides3. Esta nomenclatura aparece por primera vez en la clasificación de la OMS de 2008, en el grupo de las leucemias agudas mieloides y neoplasias de precursores relacionados, y corresponde a diversos cuadros previamente descritos con una nomenclatura antigua (linfoma NK-blástico, leucemia NK CD4+ agranular, linfoma/leucemia blástica de células NK, neoplasia hematodérmica agranular CD4+, CD56+)4.
Desde el punto de vista epidemiológico representa una entidad rara (menos del 1% de todas las leucemias agudas). Está descrito un predominio masculino (relación varón/mujer: 3/1), y una mediana de edad de 61 a 67 años, muy similar a lo observado en nuestros pacientes, aunque también hay casos publicados en la población pediátrica. Su etiología es desconocida, aunque en algunos casos, como en nuestro primer paciente, se asocia a mielodisplasia5.
Desde el punto de vista dermatológico resulta destacable que en hasta en el 80-100% de los pacientes la afectación cutánea es el primer signo de la enfermedad, asociándose frecuentemente a la afectación de la médula ósea (80%), de sangre periférica (60%), linfadenopatías y esplenomegalia (60%), siendo menos frecuente la del sistema nervioso central, que es mayor en las recaídas de la enfermedad6.
La clínica cutánea suelen consistir en nódulos únicos o múltiples, localizados en el tórax y/o las extremidades, que evolucionan en semanas o meses y se asocian generalmente a enfermedad extracutánea generalizada, como linfadenopatías, hepatoesplenomegalia, infiltración de la médula ósea, aunque cualquier órgano puede encontrarse afectado7.
La histopatología característica corresponde a un infiltrado tumoral homogéneo y monótono, con mitosis ocasionales, sin infiltrado de células reactivas con afectación dérmica e hipodérmica, sin afectar la epidermis (zona Grenz). Las células muestran citoplasmas basófilos, no muy abundantes y núcleos de tamaño medio redondos u ovalados, con cromatina finamente granular y escasos nucleolos (apariencia blástica). Los estudios inmuhistoquímicos son esenciales para el diagnóstico8. Las células neoplásicas son positivas para CD4, CD56, CD123 (receptor de IL3), CD45RA, TCL1 (protooncogén expresado en algunos linfomas B, linfocitos T inmaduros y leucemia prolinfocítica T), Bcl2, CD43, CD101, HLA-DR y Blood Dendritic cell Antigen-2, y pueden ser positivas o negativas para CD2, CD7, CD68 y TdT. Tanto los marcadores de estirpe mieloide (CD13, CD33 y mieloperoxidasa) como los de serie linfoide (CD3, CD8, CD20) son negativos, así como CD34, CD117 (c-kit) y virus de Epstein Barr9.
Los estudios de genética molecular suelen revelar una configuración de línea germinal de los genes del receptor de las células T (TCR), y de la cadena pesada de las inmunoglobulinas (IGH), con casos excepcionales de reordenamiento del TCR. Se han descrito multitud de cariotipos complejos y anomalías cromosómicas que afectan a múltiples cromosomas (5q, 6p,12p,13q,15q, y 9)9.
El pronóstico de estos pacientes es muy malo sin tratamiento, con una mediana de supervivencia de 12-14 meses con quimioterapia intensiva. Aunque hasta el 80-90% responden a quimioterapia intensiva, la inmensa mayoría (90%) recae. La supervivencia descrita es del 50% al año y del 25% a los 2 años. El trasplante alogénico de progenitores hematopoyéticos en la primera remisión completa es la única alternativa para la curación10. Se ha publicado que la edad menor a 40 años y la expresión de TdT mayor al 50% son variables independientes, asociadas con un mejor pronóstico11. También hay casos aislados de tratamiento exitoso con rituximab12.
En conclusión, presentamos 2 casos clínicos típicos de una neoplasia infrecuente, que supone menos del 1% de todas las leucemias agudas. La inmunohistoquímica es característica e indispensable para el diagnóstico. En la inmensa mayoría de los casos hay afectación cutánea inicial, que puede permitir una sospecha clínica precoz, esencial para el adecuado manejo diagnóstico-terapéutico, dado el mal pronóstico que conlleva sin tratamiento.