




Varias enfermedades autoinmunes, infecciosas, inflamatorias, genéticas o metabólicas se presentan con lesiones en la piel y las mucosas; estas se denominan en general enfermedades mucocutáneas (EMC). Las lesiones en la cavidad oral pueden ser la manifestación inicial de estas entidades o comprometerla en algún punto de su evolución. En general son dolorosas y un motivo de consulta frecuente para diversas especialidades médicas y odontológicas. Es importante conocer y diferenciar las EMC que afectan a la boca, ya que pueden ser enfermedades graves y potencialmente letales1. En esta oportunidad revisaremos la etiología, epidemiología y características clínicas más relevantes del pénfigo vulgar (PV) y de la enfermedad de Behçet (EB), además describiremos generalidades y puntos clave del manejo terapéutico.
Pénfigo vulgarEtiologíaEl pénfigo engloba un grupo de enfermedades ampollares autoinmunes potencialmente letales, caracterizadas por ampollas flácidas y erosiones en las membranas mucosas y la piel2. De etiología desconocida, tendría base poligenética, ligada a distintas moléculas HLA clase ii3. Estas participarían en la presentación de péptidos derivados de los desmosomas a clones específicos de linfocitos T CD4+secretores de citoquinas Th2 (IL4, IL6 e IL10), mediando la producción de los autoanticuerpos IgG contra moléculas de unión intercelular, como las desmogleínas (Dsg 1 y Dsg 3), proteínas de adhesión de los desmosomas de los queratinocitos epidermales, produciendo la pérdida de unión entre estos4. La expresión de desmogleínas intraepiteliales es diferente en la piel y en las mucosas. En la piel predomina la Dsg 1 y en las membranas mucosas la Dsg 3 en todo el epitelio escamoso. El perfil de anticuerpos se corresponde con las características clínicas de cada tipo de pénfigo. El PV es la variante más severa y frecuente, con 3 presentaciones clínicas: predominio mucoso, que es el más frecuente (62%) y se caracteriza por ampollas en la mucosa oral (con anticuerpos anti Dsg 3>Dsg 1), tipo mucocutáneo, que compromete tanto la epidermis como la mucosa oral (anticuerpos anti Dsg 3=Dsg 1) y predominio cutáneo, el menos frecuente (11%), con ampollas en las capas profundas de la epidermis (anticuerpos anti Dsg 1>Dsg 3)5. Aparte, se ha mencionado la influencia de diversos factores predisponentes: ambientales (dieta, estrés físico y psicológico), infecciosos (virus herpes, Epstein-Barr, citomegalovirus), fármacos con grupos tiol (penicilamina, captopril, piroxicam) y diversos tumores3.
EpidemiologíaEl PV tiene una incidencia estimada de 0,5 a 3,2 casos por 100.000, afectando a hombres y mujeres por igual. Se presenta principalmente entre la 4.ª y la 6.ª décadas de la vida y raramente compromete a niños y ancianos2. Los estudios epidemiológicos muestran mayor número de casos en ciertos grupos étnicos de origen mediterráneo, del sur de Asia y en judíos asquenazíes5,6. Antes del advenimiento de la terapia sistémica con corticoides e inmunosupresores, la mortalidad alcanzaba el 50% a los 2 años, llegando al 100% a los 5 años, fundamentalmente por infecciones y malnutrición4. Actualmente ha caído al 10% con los últimos tratamientos, siendo la inmunosupresión la responsable de la morbilidad, por el riesgo aumentado de infecciones graves. Por otro lado, el PV puede estar asociado a otras enfermedades autoinmunes como la artritis reumatoide (AR), miastenia gravis, lupus eritematoso y anemia perniciosa7.
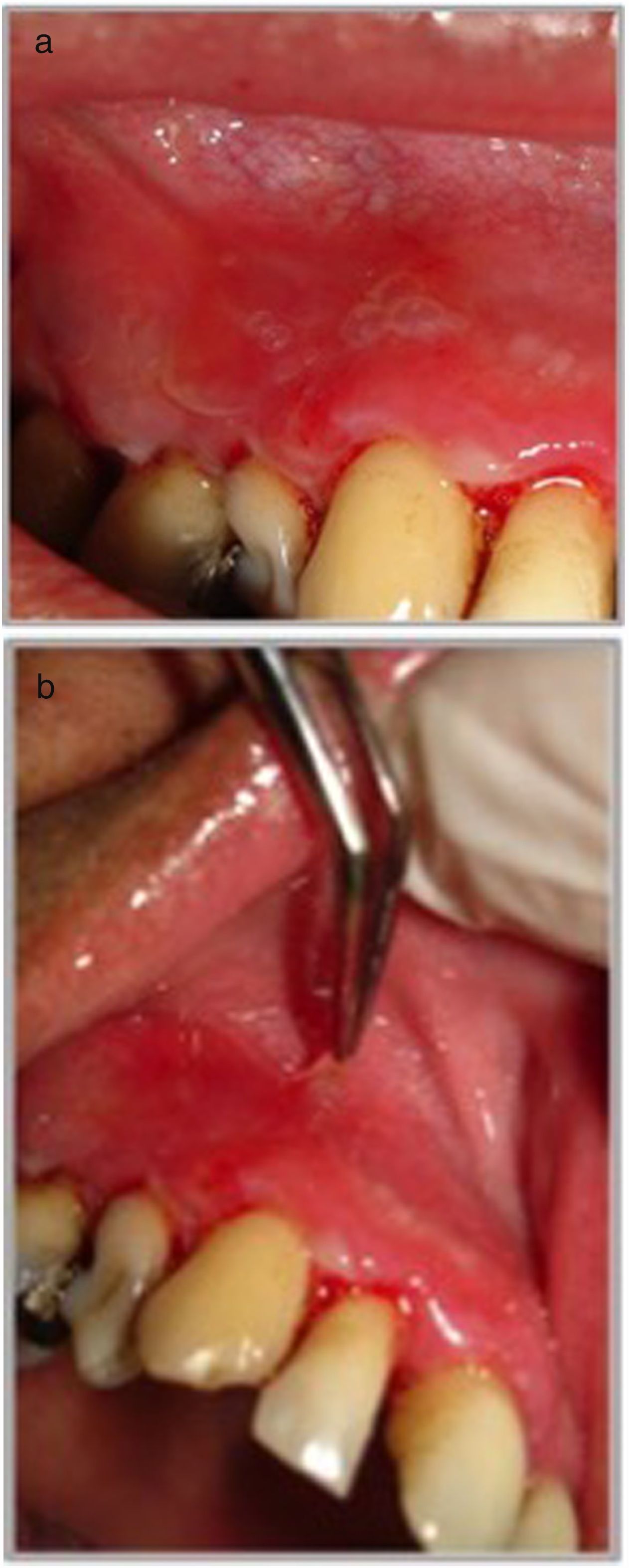
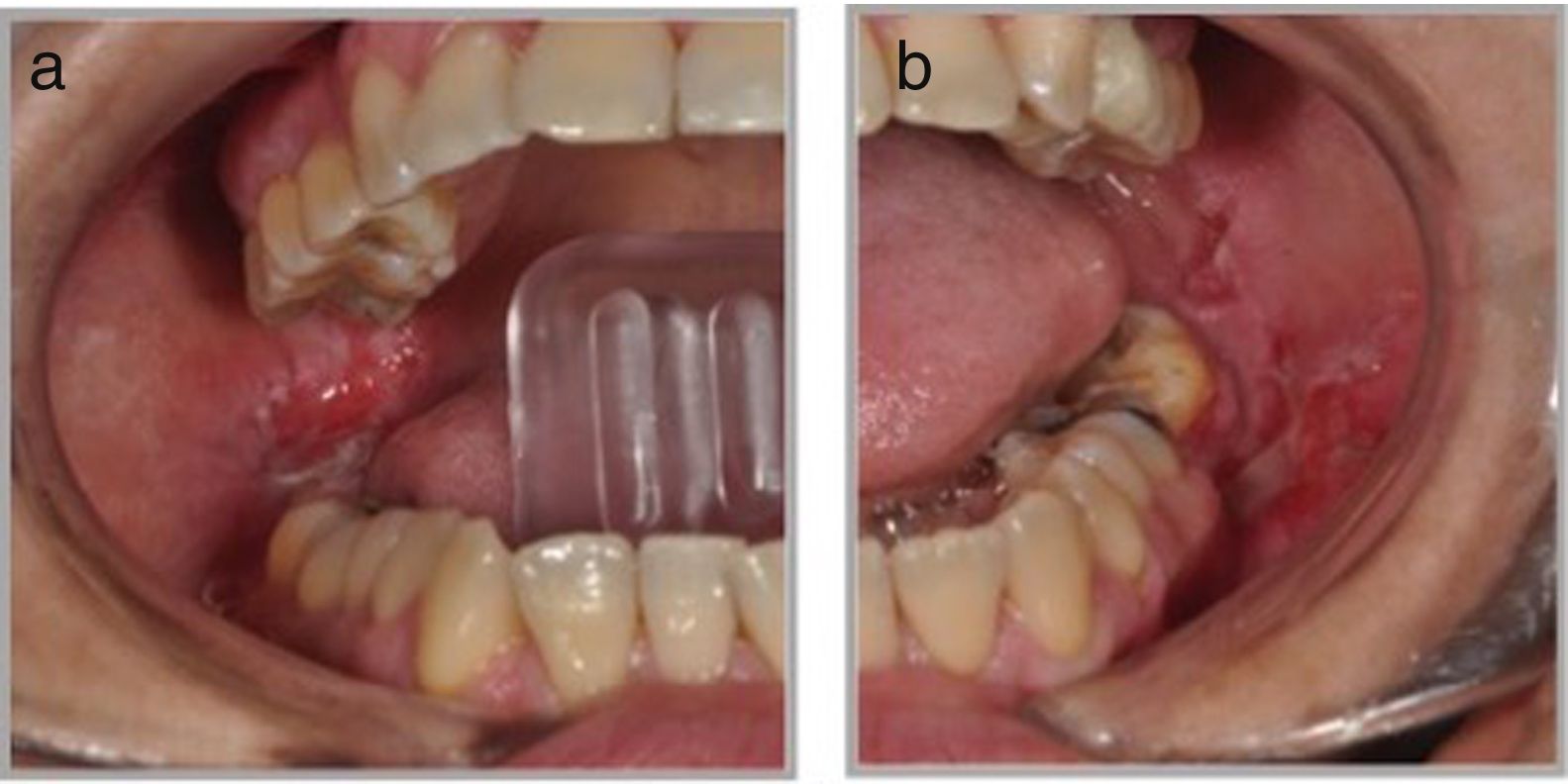
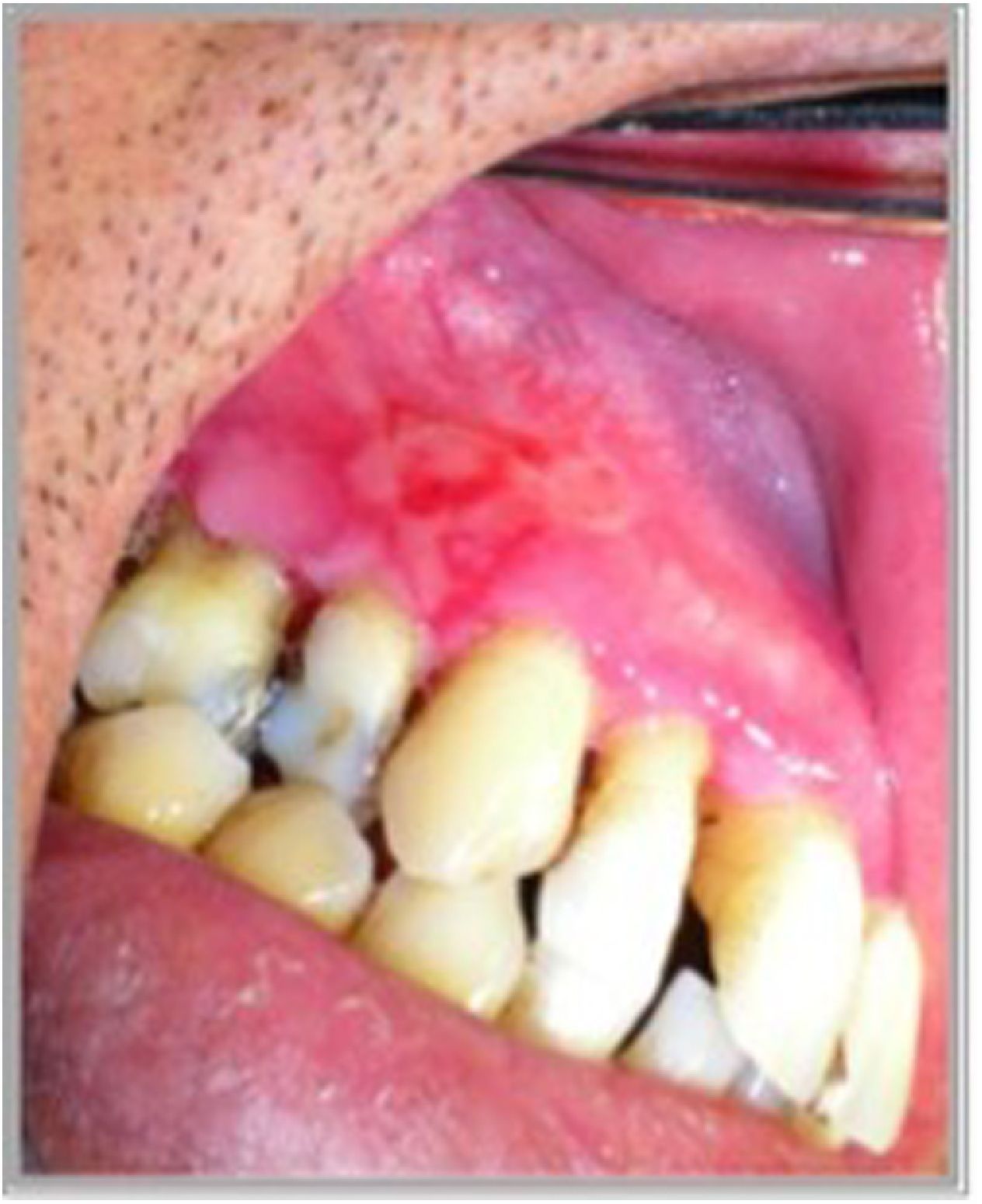
Presentación clínicaManifestaciones orales: el compromiso oral es la primera manifestación en el 50-70% de los casos y ocurre en el 90% de los pacientes durante el curso de la enfermedad1. Las lesiones elementales son múltiples ampollas flácidas, de diferentes tamaños y que característicamente se rompen al menor traumatismo o presión (signo de Nikolsky positivo), dejando una erosión muy dolorosa, de bordes irregulares, sobre una base eritematosa o blanquecina cicatricial, cubierta por una membrana o pseudomembrana (figs. 1a y b)4. Tienen curso crónico y recurrente, con lesiones en distintas etapas de evolución, por lo que es raro ver ampollas intactas. En cavidad oral las lesiones pueden aparecer, de promedio, desde 5 meses a un año antes que en la piel, y afectan principalmente a la mucosa bucal, yugal, paladar, encías y labios (figs. 2a y b). Estos últimos pueden estar inflamados y erosionados, cubiertos por pseudomembranas con exudados secos serohemáticos. El compromiso gingival puede aparecer en etapas avanzadas de la enfermedad, manifestándose como gingivitis descamativa (GD) (fig. 3). También es frecuente observar lesiones erosivas en la superficie de la lengua y del paladar (tabla 1)8.
. Estas ampollas se rompen a la menor presión o traumatismo, lo que se denomina signo de Nikolsky positivo, dejando una erosión sobre la base eritematosa (b).")
. Nótese la membrana amarillenta que recubre su superficie (b).")

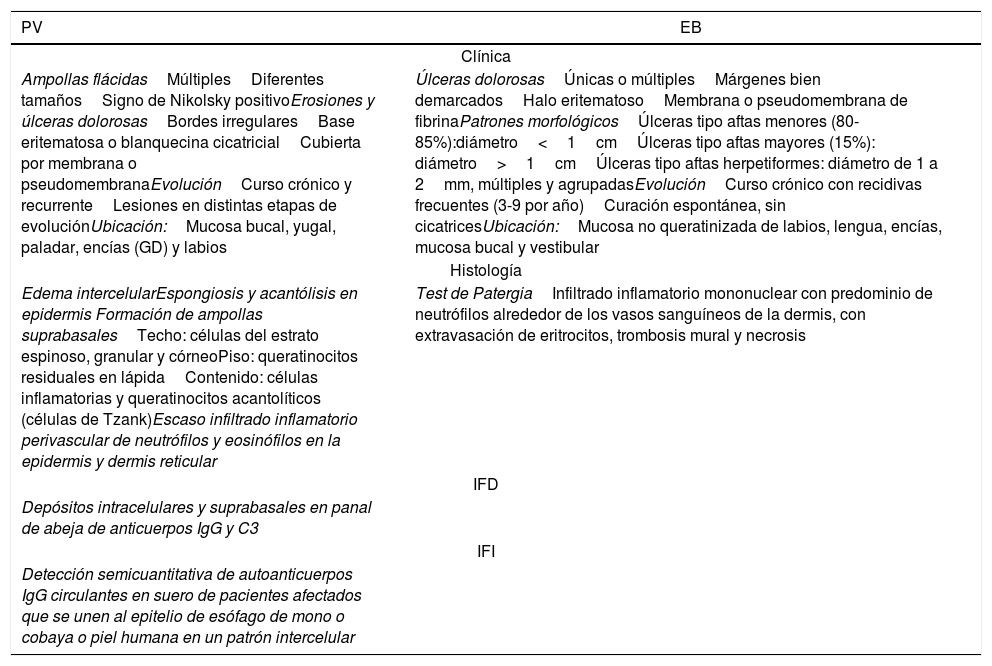
Características clínicas e inmunohistológicas del compromiso de la mucosa oral por pénfigo vulgar y enfermedad de Behçet
| PV | EB |
|---|---|
| Clínica | |
| Ampollas flácidasMúltiplesDiferentes tamañosSigno de Nikolsky positivoErosiones y úlceras dolorosasBordes irregularesBase eritematosa o blanquecina cicatricialCubierta por membrana o pseudomembranaEvoluciónCurso crónico y recurrenteLesiones en distintas etapas de evoluciónUbicación:Mucosa bucal, yugal, paladar, encías (GD) y labios | Úlceras dolorosasÚnicas o múltiplesMárgenes bien demarcadosHalo eritematosoMembrana o pseudomembrana de fibrinaPatrones morfológicosÚlceras tipo aftas menores (80-85%):diámetro<1cmÚlceras tipo aftas mayores (15%): diámetro>1cmÚlceras tipo aftas herpetiformes: diámetro de 1 a 2mm, múltiples y agrupadasEvoluciónCurso crónico con recidivas frecuentes (3-9 por año)Curación espontánea, sin cicatricesUbicación:Mucosa no queratinizada de labios, lengua, encías, mucosa bucal y vestibular |
| Histología | |
| Edema intercelularEspongiosis y acantólisis en epidermis Formación de ampollas suprabasalesTecho: células del estrato espinoso, granular y córneoPiso: queratinocitos residuales en lápidaContenido: células inflamatorias y queratinocitos acantolíticos (células de Tzank)Escaso infiltrado inflamatorio perivascular de neutrófilos y eosinófilos en la epidermis y dermis reticular | Test de PatergiaInfiltrado inflamatorio mononuclear con predominio de neutrófilos alrededor de los vasos sanguíneos de la dermis, con extravasación de eritrocitos, trombosis mural y necrosis |
| IFD | |
| Depósitos intracelulares y suprabasales en panal de abeja de anticuerpos IgG y C3 | |
| IFI | |
| Detección semicuantitativa de autoanticuerpos IgG circulantes en suero de pacientes afectados que se unen al epitelio de esófago de mono o cobaya o piel humana en un patrón intercelular | |
EB: enfermedad de Behçet; GD: gingivitis descamativa; IFD: inmunofluorescencia directa; IFI: inmunofluorescencia indirecta; PV: pénfigo vulgar.,
Manifestaciones en otras mucosas: el PV también puede comprometer la mucosa nasal (76%), la faringe (66%) y la laringe (55%), presentándose con erosiones dolorosas que dificultan la alimentación, la deglución y el habla1. En la endoscopía se puede evidenciar compromiso del esófago, del estómago, del duodeno y del ano. La afectación del esófago se ha descrito cada vez más frecuentemente, presentándose hasta en un 51% de los pacientes con lesiones en la cavidad oral que consultan por disfagia. Si bien suele ser asintomática, puede evolucionar a esofagitis disecante, con desprendimiento de pedazos de la mucosa esofágica9. El compromiso de la mucosa genital se describe hasta en el 20% de los pacientes, afectando mayormente a mujeres; se correlaciona con la severidad y la extensión del compromiso de otras mucosas, en especial la nasal10. Puede presentarse con erosiones en el periné, la vagina, el cuello cervical y con menor frecuencia en el pene11. El compromiso ocular es raro (14,3%) y se presenta asociado a otras lesiones mucocutáneas12. Se describe como una conjuntivitis bilateral que no cicatriza, o como lesiones ulceradas extremadamente dolorosas que tienden a progresar a la fibrosis, pero que no afectan a la visión13.
Manifestaciones cutáneas: el compromiso cutáneo puede ser localizado o generalizado, principalmente en el tronco, las ingles, las axilas y las áreas seborreicas, respetando la superficie palmoplantar. La mayoría de los pacientes desarrolla ampollas flácidas de contenido claro sobre piel normal o eritematosa, con signo de Nikolsky positivo, generando erosiones y úlceras que pueden desarrollar costras serohemáticas confluentes. La lesiones cicatrizan ad integrum muy lentamente, pero pueden dejar cambios de pigmentación. En cuero cabelludo se observan placas eritematosas con escamas o erosiones con costras, que pueden progresar a alopecia cicatricial14.
DiagnósticoEl diagnóstico de certeza puede ser desafiante, y requiere no solo de la sospecha clínica por las características de las lesiones en la cavidad oral, sino que muchas veces será fundamental una biopsia del tejido perilesional para análisis histológico, inmunohistoquímico y serológico5. La histología muestra edema intercelular, espongiosis y acantólisis en la epidermis, que progresa a la formación de ampollas suprabasales. El piso de las ampollas está compuesto por queratinocitos residuales que se disponen en fila sobre la membrana basal, dando un efecto denominado «en lápida». El techo se encuentra constituido por restos de células del estrato espinoso, granular y córneo. En el interior de la ampolla se observan células inflamatorias y queratinocitos acantolíticos (células de Tzank)15. Puede haber escaso infiltrado inflamatorio perivascular de neutrófilos y eosinófilos en la epidermis y dermis reticular, producto de la pérdida de la adhesión entre las células. La inmunofluorescencia directa muestra depósitos intracelulares y suprabasales de anticuerpos IgG y, ocasionalmente, proteínas C3 del complemento, con un patrón similar a un panal de abejas16. La inmunofluorescencia indirecta tiene una sensibilidad de entre el 80-90% en PV, confirma la existencia de anticuerpos IgG circulantes en el suero de pacientes afectados2. A diferencia del penfigoide, el título de estos anticuerpos se correlaciona con la extensión y severidad de la enfermedad y también permite evaluar la respuesta al tratamiento. Existen otras técnicas de laboratorio que están en constante desarrollo para el diagnóstico diferencial de PV, como el test de ELISA, que detecta anticuerpos antidesmogleína circulantes usando antígenos recombinantes, con una sensibilidad y especificidad muy altas (96% y 100%, respectivamente)17. Por otro lado, el inmunoblot y la inmunoprecipitación tienen como fundamento la detección de antígenos o regiones antigénicas específicas de células epidérmicas, usando autoanticuerpos circulantes marcados, pero tienen mayor limitación de acceso por dificultades técnicas y de precio (tabla 1)18.
Diagnóstico diferencialMuchos tipos de pénfigo presentan ampollas subepidérmicas y autoanticuerpos contra distintos componentes estructurales de la unión dermoepidérmica, por lo que el diagnóstico diferencial suele ser difícil, llegando a requerir de múltiples y costosos estudios de laboratorio2. Dentro de los subtipos de pénfigo es importante destacar el pénfigo paraneoplásico (PPN), que aunque infrecuente, produce gran compromiso de la cavidad oral7. El PPN es un síndrome paraneoplásico autoinmune de afectación multiorgánica; se asocia al linfoma no Hodgkin, a la leucemia linfocítica crónica, a la enfermedad de Castelman y al timoma13. Hay desarrollo de autoanticuerpos IgG contra varios tipos de proteínas estructurales de la membrana basal, como las desmoplaquinas, las envoplaquinas, las periplaquinas y el antígeno i del penfigoide bulloso (BP230); además, parte del daño de esta enfermedad se produce mediado por citotoxicidad directa5,16. Las lesiones pueden ser ampollares, morbiliformes o liquenoides en las mucosas y en la piel, con erosiones y úlceras difusas, dolorosas y persistentes, con un gran componente necrótico, característicamente en los bordes laterales de la lengua y en el bermellón de los labios (estomatitis erosiva intratable). La histología de las lesiones muestra abundante necrosis de queratinocitos, con degeneración vacuolar. En la inmunofluorescencia directa se observan depósitos de IgG y complemento en la membrana basal, a diferencia del PV. En general, el PPN es refractario al tratamiento inmunosupresor y el pronóstico no depende de la neoplasia asociada; es más, el tratamiento de la neoplasia primaria no afecta la actividad de la enfermedad autoinmune, pudiendo seguir cursos independientes (tabla 2)1,5.
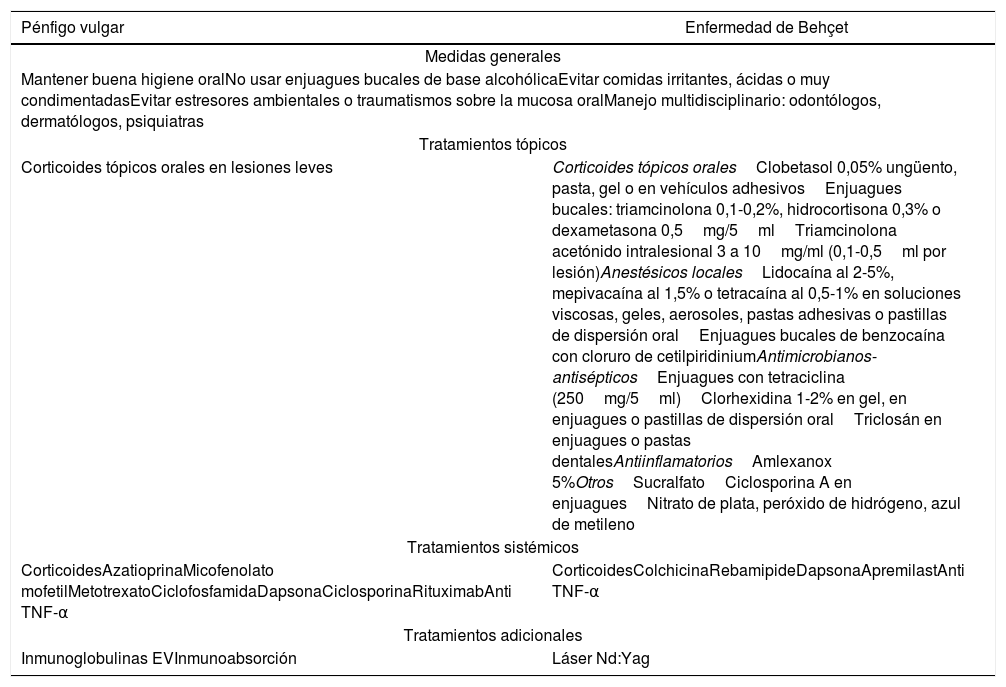
Resumen de medidas generales y manejo médico
| Pénfigo vulgar | Enfermedad de Behçet |
|---|---|
| Medidas generales | |
| Mantener buena higiene oralNo usar enjuagues bucales de base alcohólicaEvitar comidas irritantes, ácidas o muy condimentadasEvitar estresores ambientales o traumatismos sobre la mucosa oralManejo multidisciplinario: odontólogos, dermatólogos, psiquiatras | |
| Tratamientos tópicos | |
| Corticoides tópicos orales en lesiones leves | Corticoides tópicos oralesClobetasol 0,05% ungüento, pasta, gel o en vehículos adhesivosEnjuagues bucales: triamcinolona 0,1-0,2%, hidrocortisona 0,3% o dexametasona 0,5mg/5mlTriamcinolona acetónido intralesional 3 a 10mg/ml (0,1-0,5ml por lesión)Anestésicos localesLidocaína al 2-5%, mepivacaína al 1,5% o tetracaína al 0,5-1% en soluciones viscosas, geles, aerosoles, pastas adhesivas o pastillas de dispersión oralEnjuagues bucales de benzocaína con cloruro de cetilpiridiniumAntimicrobianos-antisépticosEnjuagues con tetraciclina (250mg/5ml)Clorhexidina 1-2% en gel, en enjuagues o pastillas de dispersión oralTriclosán en enjuagues o pastas dentalesAntiinflamatoriosAmlexanox 5%OtrosSucralfatoCiclosporina A en enjuaguesNitrato de plata, peróxido de hidrógeno, azul de metileno |
| Tratamientos sistémicos | |
| CorticoidesAzatioprinaMicofenolato mofetilMetotrexatoCiclofosfamidaDapsonaCiclosporinaRituximabAnti TNF-α | CorticoidesColchicinaRebamipideDapsonaApremilastAnti TNF-α |
| Tratamientos adicionales | |
| Inmunoglobulinas EVInmunoabsorción | Láser Nd:Yag |
EV: endovenoso; GD: gingivitis descamativa; VO: vía oral.
El pronóstico del PV es malo sin tratamiento, por ende este debe iniciarse sin demoras. La base son los corticoides sistémicos, orales o endovenosos, en pulsos cortos. En general, se utiliza prednisona o prednisolona a dosis de 0,5-1,5mg/kg/día vía oral, durante 2 a 3 semanas, disminuyéndola escalonadamente a dosis mínimas de 5-10mg/día de mantenimiento, según respuesta. Se puede asociar corticoides tópicos orales como adyuvante en lesiones leves; ya sea en enjuagues, geles o ungüentos. Los corticoides sistémicos producen rápido control de la enfermedad al aumentar la síntesis y vida media de desmogleínas y otras proteínas de la unión intercelular19.
Anticuerpos monoclonales anti CD20El rituximab fue aprobado por la FDA para el tratamiento del PV en 2018. Este anticuerpo monoclonal actúa bloqueando una proteína transmembrana del linfocito B, denominada CD20, impidiendo su activación y, con ello, disminuyendo los linfocitos circulantes y la producción de autoanticuerpos IgG. Está indicado como primera línea de tratamiento en PV moderado o severo de reciente comienzo, que no ha remitido con corticoides sistémicos y/o algún otro adyuvante inmunosupresor. Se utiliza endovenoso en dosis de 1.000mg por 2 veces, separados cada 15 días. Se puede repetir en dosis de 500 a 1.000mg en casos de recidivas, a los 6 o 12 meses de la primera dosis. Puede producir remisión total de la enfermedad en periodo de 6 a 9 meses. Tiene gran efecto ahorrador de corticoides al usarse asociado a estos, pero existe riesgo potencial de infecciones por inmunosupresión, en especial si se administran altas dosis de corticoides u otros inmunosupresores. Se han descrito casos de infecciones oportunistas por Pneumocystis jiroveci, citomegalovirus o reactivación del virus de la hepatitis B, entre otras20.
Inmunosupresores ahorradores de corticoidesEn caso de mala respuesta, intolerancia o efectos adversos a los corticoides, se han utilizado otros inmunosupresores, solos o como coadyuvantes. Es el caso de la azatioprina; si bien su mecanismo de acción no está del todo claro, participaría mediada por metabolitos activos, inhibiendo la síntesis de adenosina y guanina, bloqueando la producción de ADN, ARN y proteínas e inhibiendo la mitosis en los linfocitos T, B, células de Langerhans y monocitos. De forma similar el micofenolato mofetil, con una acción más específica, también interfiere en la proliferación y función, principalmente de células B y T, afectando la inmunidad celular y humoral, probando ser efectivo incluso en población pediátrica. Como tercera línea se ha utilizado, aunque en menor frecuencia, metotrexato y ciclofosfamida, esta última con importantes efectos adversos reportados como vómitos, diarrea, alopecia, infertilidad e incluso cistitis hemorrágica21.
Otros tratamientosSe describe el uso de gammaglobulinas endovenosas en altas dosis, que si bien no aumentan el riesgo de infecciones como otras terapias inmunosupresoras, se han utilizado en casos refractarios a otros tratamientos7. Tiene efectos antiinflamatorios al bloquear el receptor Fc en macrófagos y células efectoras, induciendo citoquinas antiinflamatorias y efectos inmunomoduladores, regulando la producción de anticuerpos por linfocitos B e inhibiendo la proliferación linfocitaria, lo que incrementa la respuesta a los corticoides en casos de PV recalcitrante20. Otra indicación descrita es frente a compromiso del esófago, la conjuntiva o mala respuesta a tratamiento inmunosupresor biasociado por más de 3 meses. Dado que puede aumentar la viscosidad de la sangre, la producción de fibrinógeno y el recuento de plaquetas, se han reportado casos de infartos cardíacos y tromboembolias pulmonares. También se han descrito meningitis aséptica y daño renal, por lo que se recomienda una selección cuidadosa y monitorización frecuente en pacientes que lo requieran5. Otra intervención descrita en casos graves es la inmunoabsorción, que es una plasmaféresis modificada que solo remueve IgG del plasma. Está indicada en situaciones de emergencia, en pacientes que tienen compromiso de más del 30% de la superficie corporal o más del 25% de la superficie de membranas mucosas2. Actualmente siguen en constante desarrollo nuevas terapias orientadas a modular la respuesta inmunológica y la interacción entre linfocitos T y B autorreactivos, fundamentales en la patogenia de esta grave enfermedad20,21.
Enfermedad de BehçetEtiologíaEnfermedad autoinflamatoria, caracterizada por una vasculitis multisistémica. De etiología desconocida, sería desencadenada por factores exógenos en individuos susceptibles. Tiene un curso crónico recurrente, con periodos de remisión y recaídas frecuentes, afectando a cualquier área del cuerpo. Básicamente se presenta con la tríada de úlceras orales, úlceras genitales y uveítis, pero puede tener compromiso articular, gastrointestinal, neurológico, urogenital, pulmonar y/o cardíaco22. Se sabe que habría una fuerte predisposición genética ligada a moléculas HLA-B51/B5, que desempeñarían un rol importante en la activación de neutrófilos, junto con una sobreexpresión de citoquinas proinflamatorias, principalmente dependientes de linfocitos T desregulados. Diversos factores patogénicos desencadenarían estas respuestas, como infecciones virales (herpes simplex 1, Epstein-Barr, varicela zoster, parvovirus B19, citomegalovirus y hepatitis A, B, C, E y G), antígenos bacterianos (de Helicobacter pylori, Borrelia burgdorferi o Mycoplasma fermentans) o traumatismos previos sobre la mucosa oral23. Además, se cree que habría una alteración en la ecología bacteriana oral, con aumento de especies atípicas de Streptococos, como el S. sanguinis. Esta sobrecarga bacteriana en pacientes susceptibles, provocaría aumento en la expresión de proteínas de shock térmico y otros péptidos, que son reconocidos por receptores de reconocimiento de patrones de las células presentadoras de antígenos de la barrera mucocutánea oral, estimulando una reacción cruzada con el tejido dañado expuesto del huésped, que lleva a la activación de la respuesta inmune innata y adaptativa. Hay aumento en la expresión de moléculas de adhesión de células endoteliales y sobreexpresión de citoquinas proinflamatorias dependientes de linfocitos T helper (Th1 y Th17) como la IL-6, IL-8, IL-17, IL-23, INF-γ y TNF-α, que potencian la cascada inflamatoria, estimulando la infiltración y activación de neutrófilos en el endotelio vascular. Estos, a su vez, secretan radicales libre y superóxidos que incrementan el daño tisular y aumentan, finalmente, la expresión del factor de crecimiento del endotelio vascular, causando daño entotelial, trombosis y vasculitis24. Es importante destacar el rol de esta microbiota oral alterada en la patogenia particular de la EB; se ha visto que pacientes con mala higiene oral o enfermedad periodontal tienen mayor recurrencia y severidad de brotes de úlceras orales. Del mismo modo, el uso de antibióticos o antisépticos orales en estos casos ha mostrado control de la enfermedad25. Por otro lado, existirían otros factores participando en la etiopatogenia; se han encontrado diversos tipos de complejos inmunes circulantes, sin poder precisar aún la naturaleza de los antígenos. En la histología destaca una vasculitis oclusiva inmunomediada, con anticuerpos anti células endoteliales y otros anticuerpos contra proteínas del endotelio vascular que, de alguna manera, participarían en la pronunciada inflamación perivascular. La trombomodulina, una glucoproteína de la superficie de las células endoteliales, está aumentada en la EB, al igual que otras moléculas secretadas frente al daño del endotelio vascular, como la endotelio 1 plasmática, que sumado a una disminución de la actividad fibrinolítica y de los niveles circulantes de factor xii favorecerían mayor vasoconstricción y trombosis26,27.
EpidemiologíaAunque poco frecuente, la EB presenta un claro predominio endémico en la llamada Ruta de la Seda, principalmente en las penínsulas de Corea y Japón y en países de la costa mediterránea. Tiene una prevalencia variable en esta zona de entre 14-20 casos por 100.000; destaca Turquía con hasta 80 casos por 100.000, siendo mucho menos frecuente en el resto del mundo (Estados Unidos: 5,2 casos por 100.00028). Afecta en proporción similar a hombres y mujeres, aunque hay más hombres afectados en población de origen árabe y más mujeres afectadas en Japón y Corea. El género también pareciera influenciar el pronóstico, ya que los hombres presentan cuadros más graves, con mayor compromiso ocular, lesiones pápulo-pustulares y trombosis venosas profundas y superficiales, mientras que en las mujeres las lesiones son menos intensas, con mayor compromiso genital y articular. Se presenta, en promedio, entre los 20 y los 35 años; mientras más precoz es el inicio de las manifestaciones, peor es el pronóstico, con el consecuente aumento de la morbimortalidad24,27.
Presentación clínicaSi bien puede haber compromiso multisistémico, las lesiones mucocutáneas son el sello distintivo, presentándose desde el inicio o desarrollándose con el curso de la enfermedad. Las úlceras orales aftosas están en más del 80% de los pacientes, precediendo el compromiso en otras regiones corporales, en promedio unos 7-8 años. Le siguen en frecuencia las úlceras genitales recurrentes y las lesiones cutáneas. De forma más variable, puede haber manifestaciones por vasculitis en otros órganos como uveítis, mono o poliartritis seronegativas, trombosis arterial o venosa, aneurismas, pericarditis o endocarditis y también compromiso gastrointestinal, renal y pulmonar1,29.
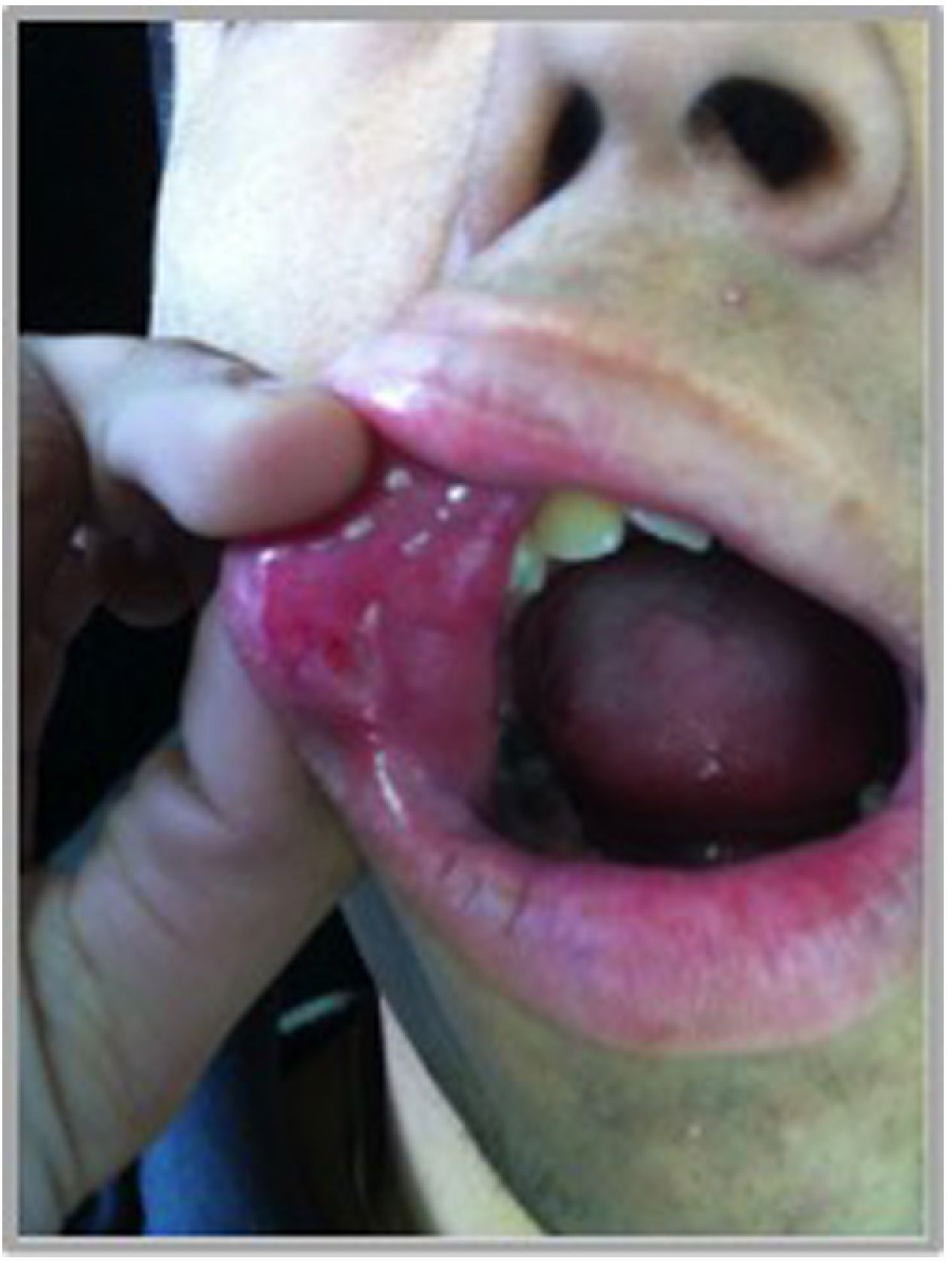
Manifestaciones orales: las úlceras orales son muy dolorosas, pueden ser únicas o múltiples, con márgenes bien demarcados, un halo eritematoso y están cubiertas por una membrana o pseudomembrana amarillenta o grisácea de fibrina. Se ubican de preferencia en mucosa no queratinizada y de localización anterior (labios, lengua, encías, mucosa bucal y vestibular). Se describen 3 patrones morfológicos: úlceras tipo aftas menores, con un diámetro inferior a 1cm (80-85%) (fig. 4); úlceras tipo aftas mayores, con un diámetro mayor a 1cm (15%) y las menos frecuentes, úlceras tipo aftas herpetiformes, múltiples úlceras agrupadas, con un diámetro de 1 a 2mm23. Las lesiones más pequeñas duran en promedio 7 a 10 días, mientras que úlceras más grandes pueden tardar hasta 4 semanas en cicatrizar. Si bien evolucionan rápidamente hacia la curación espontánea, sin dejar cicatrices, las recidivas son frecuentes, con una media de 3 a 9 brotes por año. El principal problema que generan estas lesiones es la dificultad para hablar, tragar o comer por dolor (tabla 1)8,30.

Se observa una úlcera única tipo aftas menores en un paciente con EB. Característicamente presenta un diámetro inferior a 1cm, ubicada en la mucosa no queratinizada del labio superior. Se pueden observar márgenes bien demarcados, con base eritematosa y restos de fibrina en su interior.
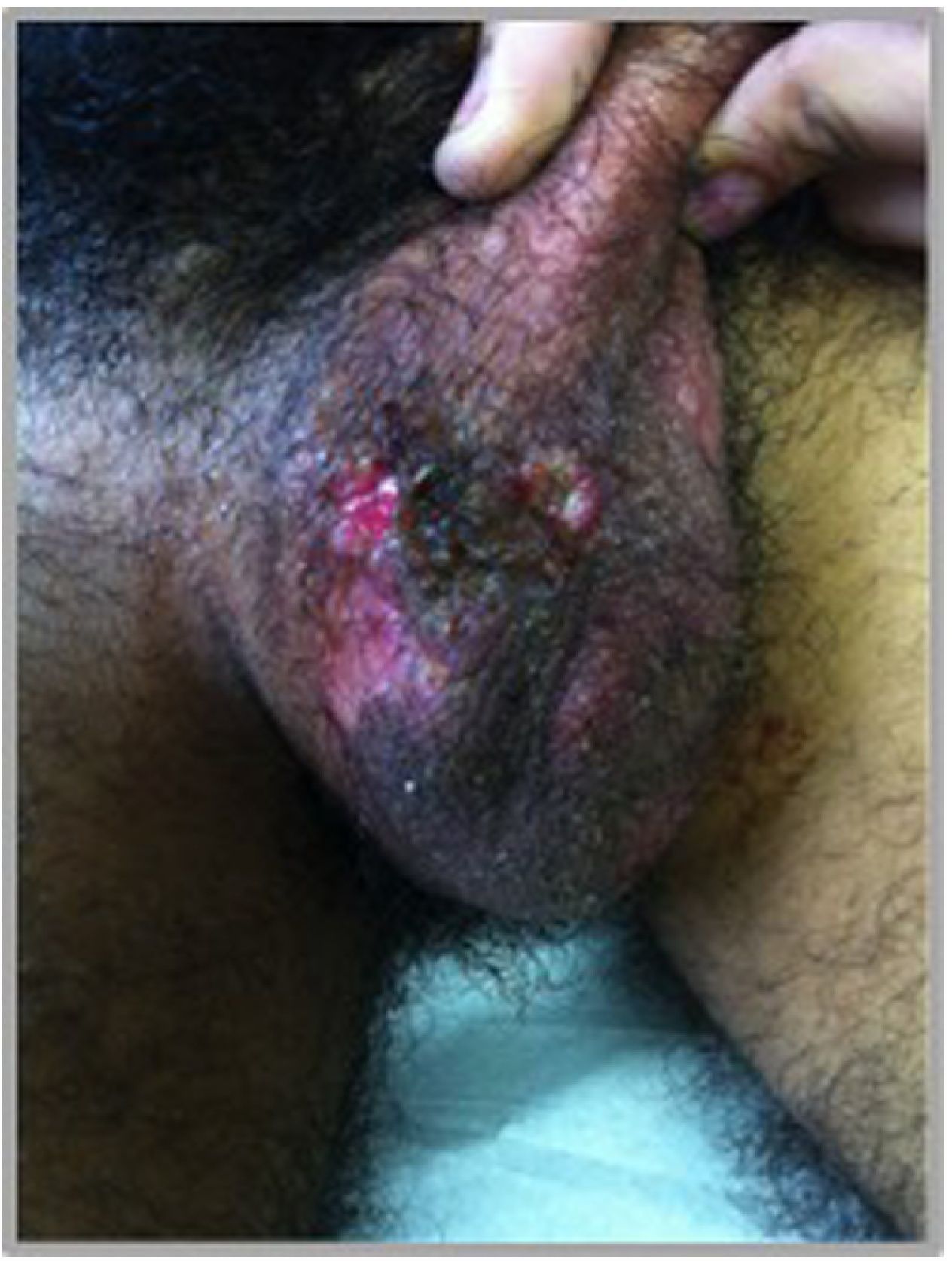
Manifestaciones en otras mucosas: el compromiso de la mucosa genital es muy frecuente, afectando a más del 60% de los pacientes27. Las lesiones tienen similares características a las orales, pero las úlceras suelen ser más grandes y profundas, curan más lentamente, recidivan menos, pero dejan cicatrices. En las mujeres las zonas más afectadas son los labios mayores y en los hombres el escroto, pero puede comprometer también otras áreas de la región perineal y perianal (fig. 5). Se han reportado úlceras en la uretra y la vejiga que pueden progresar y producir fístulas25. El compromiso ocular se reporta en más del 70% de los pacientes; si bien no hay afectación de la mucosa conjuntival, el daño se produce por vasculitis y se manifiesta básicamente como panuveítis, vasculitis retinal y queratitis31.

Manifestaciones cutáneas: las lesiones cutáneas no son específicas de EB, pero los hallazgos más frecuentes son pápulas y pústulas que se asemejan al acné vulgar, pero sin comedones. También pueden aparecer lesiones nodulares tipo eritema nodoso hasta en un tercio de los pacientes, principalmente en las extremidades inferiores, pero también en la cara, en los brazos y en los glúteos. Las tromboflebitis superficiales y las lesiones vasculíticas son lesiones nodulares que se agrupan de forma lineal, como cordones eritematosos y dolorosos, ubicados principalmente en el tronco y en las extremidades25.
DiagnósticoNo existe prueba de laboratorio específica o características histológicas patognomónicas de la EB, por lo que el diagnóstico es fundamentalmente clínico. Varios grupos internacionales de estudio de la EB han establecido criterios diagnósticos. En general, consideran fundamentales las úlceras orales y genitales recurrentes y las lesiones oculares; el resto de las manifestaciones sistémicas y el test de patergia (TP) son criterios menos relevantes31. El fenómeno denominado patergia cutánea consiste en una reacción inflamatoria de la piel inducida por traumatismos mínimos, que al producir disrupción epidérmica desencadenan la aparición de una pápula o pústula en la zona lesionada pasadas 24-48horas32. Si bien el análisis histológico de esta reacción tendría alta especificidad para EB (98,4%), su sensibilidad va disminuyendo según el origen de la población (68% en marroquíes, 57,4% en iraníes, 56% en turcos, 44% en japoneses, 40% en coreanos, 32% en británicos y alrededor de 20% en estadounidenses)28,33,34. De igual manera, la incidencia del TP positivo ha disminuido progresivamente en el tiempo; se desconoce la causa, pero la alta especificidad del test aún permite utilizarlo para el diagnóstico de la enfermedad35. Por otro lado, el análisis histológico de las biopsias de las lesiones mucosas muestra un infiltrado inflamatorio con predominio mononuclear alrededor de los vasos sanguíneos de la dermis, con aumento del número de mastocitos y extravasación de eritrocitos, trombosis mural y necrosis; estas características pueden variar según la antigüedad de la lesión (tabla 1)27.
Diagnóstico diferencialPara poder hacer el diagnóstico diferencial de EB hay que distinguirla de otras enfermedades que también presentan úlceras orales tipo aftas, principalmente la estomatitis aftosa recurrente, que constituye una de las entidades más frecuentes, con una incidencia de entre el 5% al 25% de población36. En general, más de 6 úlceras orales aftosas de tamaño variable, sobre base eritematosa, con compromiso del paladar blando y orofaringe, más la presencia de lesiones en otras mucosas o la afectación multisistémica y otros signos de la vasculitis, orientan a EB27. Las úlceras orales complejas pueden ser manifestaciones de infecciones (virus herpes, HIV y sífilis), alergias alimentarias (gluten, proteínas de la leche de vaca, preservantes y colorantes en alimentos, etc.), deficiencias nutricionales (vitaminas del grupo B, ácido fólico, zinc, hierro), enfermedades ampollares autoinmunes y otras EMC (liquen plano, pénfigo, penfigoide de membranas mucosas), enfermedades inflamatorias intestinales, reacciones adversas a fármacos (AINE) y otras diversas, como pioderma gangrenoso (PG), síndrome de Sweet o Neutropenia cíclica37. El primer paso para diferenciarlas es una historia clínica completa y examen físico exhaustivo, buscando compromiso de otros órganos. Según la sospecha clínica, solicitaremos exámenes de laboratorio para descartar déficit de vitaminas y minerales (hemograma con recuento diferencial, cinética del hierro, niveles plasmáticos de vitaminas y minerales, etc.), test serológicos (anticuerpos antinucleares y anticitoplasma de neutrófilos), diferentes cultivos para virus, bacterias y hongos, endoscopia digestiva o biopsias perilesionales para análisis histopatológico e inmunohistoquímico1. Por otro lado, el TP también puede ser positivo en otras enfermedades tan diversas como el PG, la leucemia mieloide crónica, AR, la enfermedad de Crohn y el herpes genital38.
TratamientoAunque no hay cura para la EB, el objetivo fundamental del tratamiento es aliviar los síntomas, resolver de forma rápida la inflamación, prevenir el daño del tejido, reducir la frecuencia y severidad de los brotes y evitar complicaciones29. Una reciente revisión del Cochrane pretendió evaluar las diversas intervenciones tópicas y sistémicas disponibles actualmente para el manejo de las úlceras orales en la EB. Se analizaron 15 trabajos publicados entre 1980 y 2012, con más de 880 pacientes tratados, sin demostrar evidencia científica de calidad suficiente para apoyar o refutar algunos de los tratamientos propuestos, por lo que la elección dependerá de la extensión y severidad de las lesiones39. Considerando el curso crónico y recurrente de la EB, el manejo es paliativo y sintomático, basado en medidas generales de alimentación e higiene oral, además de tratamientos tópicos orales y/o fármacos sistémicos, según sea el caso (tabla 2)1,27.
Medidas generalesLas lesiones en la cavidad oral son dolorosas, por ende las recomendaciones generales incluyen dieta blanda, evitando alimentos muy condimentados, irritantes, alcohol o bebidas carbonatadas. Además, el mantenimiento de una buena higiene oral diaria es fundamental, evitando el uso de enjuagues orales de base alcohólica o pastas dentales que contengan lauryl sulfato de sodio. Debiera insistirse en incentivar el abandono tabáquico, ya que se ha demostrado una disminución de las recurrencias de las úlceras aftosas23. Se ha reportado utilidad como terapia coadyuvante el ejercicio frecuente, la meditación, el yoga y otras técnicas de relajación y control de estrés, básicamente contribuyendo a la disminución de las recurrencias27.
Tratamientos tópicos oralesCorticoides oralesLos corticoides de alta o muy alta potencia se han utilizado ampliamente para tratar las manifestaciones orales de EB, fundamentalmente por el rápido control de la inflamación aguda. Clobetasol al 0,05% en ungüento, pasta, gel o en vehículos adhesivos se utiliza en úlceras pequeñas aplicado 4 veces al día de 4 a 6 semanas, solo o en combinación con antisépticos orales, como clorhexidina en gel, en enjuagues o pastillas de dispersión oral. Enjuagues bucales con corticoides 3 a 4 veces al día (triamcinolona 0,1-0,2%, hidrocortisona 0,3% o dexametasona 0,5mg/5ml) se pueden utilizar en úlceras difusas. En úlceras severas, profundas y dolorosas, triamcinolona acetónido intralesional 3 a 10mg/ml, inyectando a dosis de 0,1-0,5ml por lesión, ha mostrado utilidad. Como efectos adversos se describen ardor, alteración transitoria del gusto y candidiasis oral secundaria; puede haber absorción sistémica por uso prolongado o por deglutir los enjuagues25.
Anestésicos localesSe describe el uso de lidocaína al 2-5%, mepivacaína al 1,5% o tetracaína al 0,5-1%, en distintas preparaciones para uso en mucosas, como soluciones viscosas, geles, aerosoles, pastas adhesivas o pastillas de dispersión oral. También en preparados para enjuagues bucales de benzocaína con cloruro de cetilpiridinium36.
Antimicrobianos-antisépticosSu uso en enjuagues bucales tiene como objetivo controlar la contaminación bacteriana y la sobreinfección secundaria, además de reducir el tamaño, la duración y el dolor producido por las úlceras, pero no afectan la incidencia o la recurrencia de las lesiones. Los más usados son: la tetraciclina, que no se debe usar más de 5 días, ya que puede producir disgeusia, ardor, odinofagia, irritación en la piel, queilitis angular o candidiasis oral por disbacteriosis; el gluconato de clorhexidina al 1-2% en enjuagues 3 veces al día, que es amargo y puede teñir los dientes y la lengua; también el triclosán en enjuagues bucales o pastas dentales tiene efecto antiinflamatorio y analgésico27.
AntiinflamatoriosEl amlexanox 5% en pasta oral mucoadhesiva inhibe la formación y liberación de mediadores inflamatorios de los mastocitos y neutrófilos, acelerando la cicatrización y disminuyendo el dolor de las úlceras orales, pero sin efecto en las recurrencias. El ácido 5-aminosalicílico al 5% en crema, 3 veces al día, reduce la duración y el dolor de las lesiones26.
OtrosEl sucralfato es un complejo de sacarosa-sulfato-aluminio que tiene efectos citoprotectores al unirse a las proteínas del cráter ulceroso y membranas mucosas, formando una barrera protectora, favoreciendo la cicatrización. Se ha demostrado su efectividad en suspensiones orales en dosis de 1g/5ml, administrado 4 veces al día25. La ciclosporina A oral, en solución para enjuagues, 3 veces al día, tiene efecto inmunosupresor, con buena efectividad en reducir la duración de las lesiones37. En la literatura se mencionan otras alternativas para manejar las lesiones orales, muchas sin evidencia demostrada de utilidad, como las soluciones cáusticas de nitrato de plata, peróxido de hidrógeno y azul de metileno, entre otros23. Se ha reportado un rápido efecto analgésico y promotor de la cicatrización en las lesiones aftosas orales con el uso de láser neodimio-Yag, que es bien tolerado y no tiene efectos adversos significativos27.
Tratamientos sistémicosIndicados en casos de lesiones severas, recalcitrantes, resistentes al tratamiento tópico o con compromiso sistémico.
CorticoidesLa prednisona oral a dosis iniciales de 1mg/kg/día, una vez al día en la mañana, en pulsos cortos de una a 2 semanas, con disminución escalonada para evitar efectos adversos, es el fármaco de elección. Se indica sola o asociada a otros fármacos sistémicos ahorradores de corticoides. Actúa rápidamente controlando la inflamación y acortando la evolución de las lesiones, pero sin efecto en las recurrencias23.
ColchicinaEn dosis de 1-2mg/día, durante 4 a 6 semanas, reduce el número y duración de las úlceras al disminuir la función fagocítica de los neutrófilos. Puede producir náuseas, dolor abdominal, diarrea, cefaleas y recurrencias de las lesiones al suspender el tratamiento. Está contraindicada en el embarazo26.
DapsonaDe utilidad en úlceras orales y genitales en EB por su efecto antiinflamatorio y antibiótico, al inhibir la quimiotaxis de los neutrófilos, en dosis de 100-150mg/día, en pacientes con úlceras complejas que no responden a colchicina, aunque las recidivas al suspender el fármaco son frecuentes. Como efectos adversos se describen metahemoglobinemia, agranulocitosis, hemólisis y neuropatía periférica, por lo que requiere exámenes de laboratorio de control en forma frecuente (recuento sanguíneo completo, función renal y hepática)26.
ApremilastEs un inhibidor de la fosfodiesterasa 4, aumenta el AMP cíclico intracelular, particularmente en células del sistema inmune, disminuyendo la producción de citoquinas proinflamatorias como IL-2, IL-5, IL-13, IL-23, TNF-α e INF-γ y aumentando las citoquinas reguladoras antiinflamatorias, como IL-10. Si bien su uso en el tratamiento de úlceras orales en EB está aún en evaluación, hay varios reportes de casos con remisión exitosa de lesiones orales, en dosis de 30mg, 2 veces al día, por 12 semanas40.
Anti factor de necrosis tumoral αLa base de la patogenia de la EB es la hiperreactividad de la respuesta linfocitaria frente a diversos factores predisponentes, con la liberación de citoquinas proinflamatorias, dentro de las que destaca el TNF-α5. Se han utilizado diferentes fármacos que inhiben la acción de esta molécula, ejerciendo una función inmunomoduladora, antiangiogénica y antiinflamatoria. En ese sentido, la talidomida a dosis de 50mg/día logra la remisión de las lesiones, en especial en pacientes positivos para el VIH26. Como efectos adversos se describen neuropatía periférica, dolor abdominal y fatiga. La pentoxifilina es otro inhibidor del TNF-α, que además suprime la acción de linfocitos T CD8+citotóxicos, que están aumentados en las lesiones ulceradas23. El infliximab es un anticuerpo monoclonal quimérico anti TNF-α que se administra endovenoso, muy efectivo en el manejo de úlceras orales y genitales recurrentes; induce una rápida cicatrización sin evidencia de recurrencias al terminar el tratamiento24. El etanercept es un dímero recombinante, compuesto por una porción del dominio Fc de la IgG1 humana y por una parte del receptor del TNF-α. Actúa inhibiendo la unión de TNF-α en sus receptores celulares, reduciendo la producción de citoquinas proinflamatorias. Se administra por inyección subcutánea, 2 veces a la semana, con promisorios efectos en úlceras orales recurrentes. Tiene efectos adversos importantes y graves, como la reactivación de tuberculosis y otras infecciones, leucoencefalopatía multifocal progresiva, linfomas y reacciones tipo lupus; por ende, se aconseja administración controlada y supervisión frecuente25.