




El solapamiento clínico y bioquímico de diversas enfermedades del metabolismo fosfocálcico puede conllevar un erróneo diagnóstico y su consecuente abordaje clínico. Un ejemplo es el seudohipoparatiroidismo, que puede confundirse con el raquitismo dependiente de vitamina D (VDDR1) si no se hacen las determinaciones bioquímicas adecuadas.
Pacientes y métodosDos parejas de hermanos, de familias independientes, fueron diagnosticados clínicamente en la adolescencia de seudohipoparatiroidismo al presentar hipocalcemia, niveles elevados de hormona paratiroidea y valores normales o elevados de fósforo. Tras descartar alteraciones en GNAS, se realizó un estudio, mediante secuenciación masiva, de genes asociados a otros diagnósticos diferenciales.
ResultadosSe identificaron 2variantes genéticas en el gen CYP27B1 potencialmente asociadas con el fenotipo. Variantes patogénicas en este gen se asocian con VDDR1A. La reevaluación clínica-bioquímica de los pacientes confirmó dicho diagnóstico y se adecuó el tratamiento.
ConclusionesSi bien la VDDR1A es un trastorno del metabolismo de diagnóstico infrecuente en la edad adulta, en casos de hipocalcemia con valores elevados de PTH es relevante la determinación de las formas 1,25(OH)2D3 y 25(OH)D3 de la vitamina D para alcanzar un diagnóstico correcto.
The clinical and biochemical overlap of various pathologies of phosphocalcic metabolism can lead to misdiagnosis and consequent clinical management. One example is pseudohypoparathyroidism, which can be confused with vitamin D-dependent rickets (VDDR1) if appropriate biochemical determinations are not performed.
Patients and methodsTwo pairs of siblings, from independent families, were clinically diagnosed in adolescence with pseudohypoparathyroidism due to hypocalcaemia, elevated parathyroid hormone levels and normal or elevated phosphorus values. After ruling out alterations in GNAS, a massive sequencing study of genes associated with other differential diagnoses was carried out.
ResultsTwo genetic variants in the CYP27B1 gene potentially associated with the phenotype were identified. Pathogenic variants in this gene are associated with VDDR1A. Clinical-biochemical re-evaluation of the patients confirmed this diagnosis and treatment was adapted.
ConclusionsAlthough VDDR1A is an infrequently diagnosed pathology in adulthood, in cases of hypocalcaemia with elevated PTH values, determination of the 1,25(OH)2D3 and 25(OH)D3 forms of vitamin D is relevant to reach a correct diagnosis.
El seudohipoparatiroidismo (PHP), recientemente renombrado como iPPSD2 y 3 (del inglés inactivating PTH/PTHrP signaling disorders)1, describe un grupo heterogéneo de enfermedades genéticas o epigenéticas raras caracterizadas por la presencia de hipocalcemia e hiperfosfatemia como consecuencia de la resistencia de ciertos tejidos a las acciones biológicas de la hormona paratiroidea (PTH)2.
Sin embargo, los niveles elevados de PTH no son específicos de iPPSD, ya que existen otras condiciones fisiológicas que pueden afectarlos, como hiperparatiroidismo, tumores benignos en las glándulas paratiroideas, enfermedad renal, desórdenes en el metabolismo fosfocálcico o deficiencia de vitamina D, enfermedades que deben ser descartadas antes de dicho diagnóstico1.
Presentamos 2parejas de hermanos que, en la infancia o la adolescencia, habían sido diagnosticados de seudohipoparatiroidismo. Tras el estudio genético, en su etapa adulta, mediante secuenciación masiva de un panel de genes asociados a iPPSD y otros diagnósticos diferenciales, se identificó la alteración causal en el gen CYP27B1, lo que sugería el diagnóstico de raquitismo dependiente de vitamina D de tipo 1A.
Pacientes y métodosFamilia 1Caso 1Mujer caucásica de 59 años que se remite a consulta de asesoramiento genético por sospecha clínica de seudohipoparatiroidismo de tipo 2, diagnosticada en la infancia, por hallazgo incidental de valores elevados de PTH tras accidente con traumatismo. En la actualidad está en tratamiento con calcio y calcitriol, con PTH elevada asociada a alteraciones de la vitamina D (tabla 1). La paciente presenta una talla baja (142cm; <p3), facies redondeada, sobrepeso (61,4kg; IMC: 30,5) y discreta braquidactilia de los metacarpianos II y III (fig. A del material suplementario).
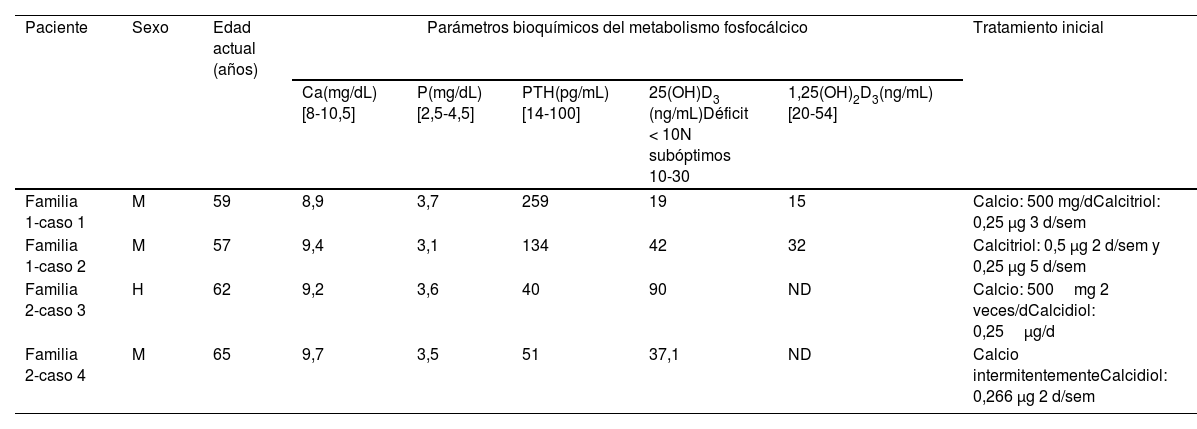
Resumen de los datos del metabolismo fosfocálcico en el momento en que los pacientes fueron a la consulta
| Paciente | Sexo | Edad actual (años) | Parámetros bioquímicos del metabolismo fosfocálcico | Tratamiento inicial | ||||
| Ca(mg/dL)[8-10,5] | P(mg/dL)[2,5-4,5] | PTH(pg/mL)[14-100] | 25(OH)D3 (ng/mL)Déficit < 10N subóptimos 10-30 | 1,25(OH)2D3(ng/mL)[20-54] | ||||
| Familia 1-caso 1 | M | 59 | 8,9 | 3,7 | 259 | 19 | 15 | Calcio: 500 mg/dCalcitriol: 0,25 μg 3 d/sem |
| Familia 1-caso 2 | M | 57 | 9,4 | 3,1 | 134 | 42 | 32 | Calcitriol: 0,5 μg 2 d/sem y 0,25 μg 5 d/sem |
| Familia 2-caso 3 | H | 62 | 9,2 | 3,6 | 40 | 90 | ND | Calcio: 500mg 2 veces/dCalcidiol: 0,25μg/d |
| Familia 2-caso 4 | M | 65 | 9,7 | 3,5 | 51 | 37,1 | ND | Calcio intermitentementeCalcidiol: 0,266 μg 2 d/sem |
Se incluye el tratamiento que estaban tomando en ese momento.
1,25(OH)2D3: niveles séricos de 1-alfa,25-dihidroxicolecalciferol o calcitriol; 25(OH)D3; niveles séricos de 25-hidroxivitamina D o calcidiol; Ca: niveles séricos de calcio; H: hombre; M: mujer; mg: miligramos; ND: no disponible; P: niveles séricos de fósforo; PTH: niveles séricos de hormona paratiroidea; μg: microgramos, se muestra entre corchetes el rango de normalidad.
Mujer de 57 años, hermana de la anterior, con sospecha clínica de PHP de tipo 2. A pesar del tratamiento, mantiene PTH elevada con niveles normales de 1,25(OH)2D3 y de 25(OH)D3 (tabla 1). Fenotípicamente, presenta una talla en el límite inferior de la normalidad (152cm; p3-p10) y sobrepeso (61,2kg; IMC: 26,5). Las radiografías muestran un ligero acortamiento de los metacarpianos II y III (fig. B del material suplementario).
Familia 2Caso 3Hombre de 62 años que acude a consulta de endocrinología diagnosticado clínicamente de seudohipoparatiroidismo a los 23 años por hipocalcemia (5,8mg/dL; rango normal 8-11mg/dL), hiperfosfatemia (5,4mg/dL; rango normal 2,7-4,5mg/dL) con niveles de PTH normales (valores no disponibles). La exploración física revela talla baja (150cm; >p3), peso normal (54,7kg; IMC: 24,31) y ausencia de osificaciones subcutáneas y de braquidactilia.
En el momento de la consulta está con tratamiento oral con calcidiol y suplementos de calcio. La analítica revela normalidad en los parámetros bioquímicos (tabla 1).
Caso 4Mujer de 65 años con sospecha clínica de seudohipoparatiroidismo familiar que acudió a consulta de endocrinología a la edad de 21 años con tetanias secundarias a hipocalcemia (5,2mg/dL), rigidez en las manos y contractura peribucal. Asimismo, se detectó acortamiento bilateral de los metacarpianos II, III y IV en radiografía (fig. C del material suplementario) y opacidades en la cápsula posterior del cristalino en ambos ojos. A lo largo del seguimiento se detectaron, ocasionalmente, niveles elevados de PTH (101 pg/ml).
La existencia del mismo cuadro en un hermano (caso 3), las características bioquímicas, y la presencia de talla baja (136cm; <p3), sobrepeso (52,5kg; IMC: 28,4) y cara redondeada conllevaron el diagnóstico de seudohipoparatiroidismo familiar y fue tratada con calcidiol (0,266μg 2días/semana) y suplementos cálcicos en presencia de tetania. La última analítica, a los 65 años, mostraba niveles séricos normales del metabolismo fosfocálcico (tabla 1).
Estudios molecularesEn ambas familias se analizaron las alteraciones epigenéticas y genéticas en el locus GNAS, tal y como se ha descrito3. A continuación, se realizó el estudio del panel NGS iPPSD-v1, de diseño propio, en el que se estudian 92 genes (ver material suplementario) asociados a iPPSDs y otros diagnósticos diferenciales. El análisis de los datos de NGS así como el filtrado y la priorización de las variantes se llevó a cabo según lo ya descrito3. Los posteriores estudios de confirmación y cosegregación se hicieron mediante secuenciación Sanger.
Todos los estudios han sido revisados y aprobados por el Comité Ético de Investigación Clínica de Euskadi (CEI-E) y, además, los pacientes han firmado el correspondiente consentimiento informado para la realización del estudio.
ResultadosTras descartar alteraciones epigenéticas y genéticas en locus GNAS, el estudio de panel NGS iPPSD-v1 permitió identificar una variante potencialmente asociada con el fenotipo en cada una de las familias.
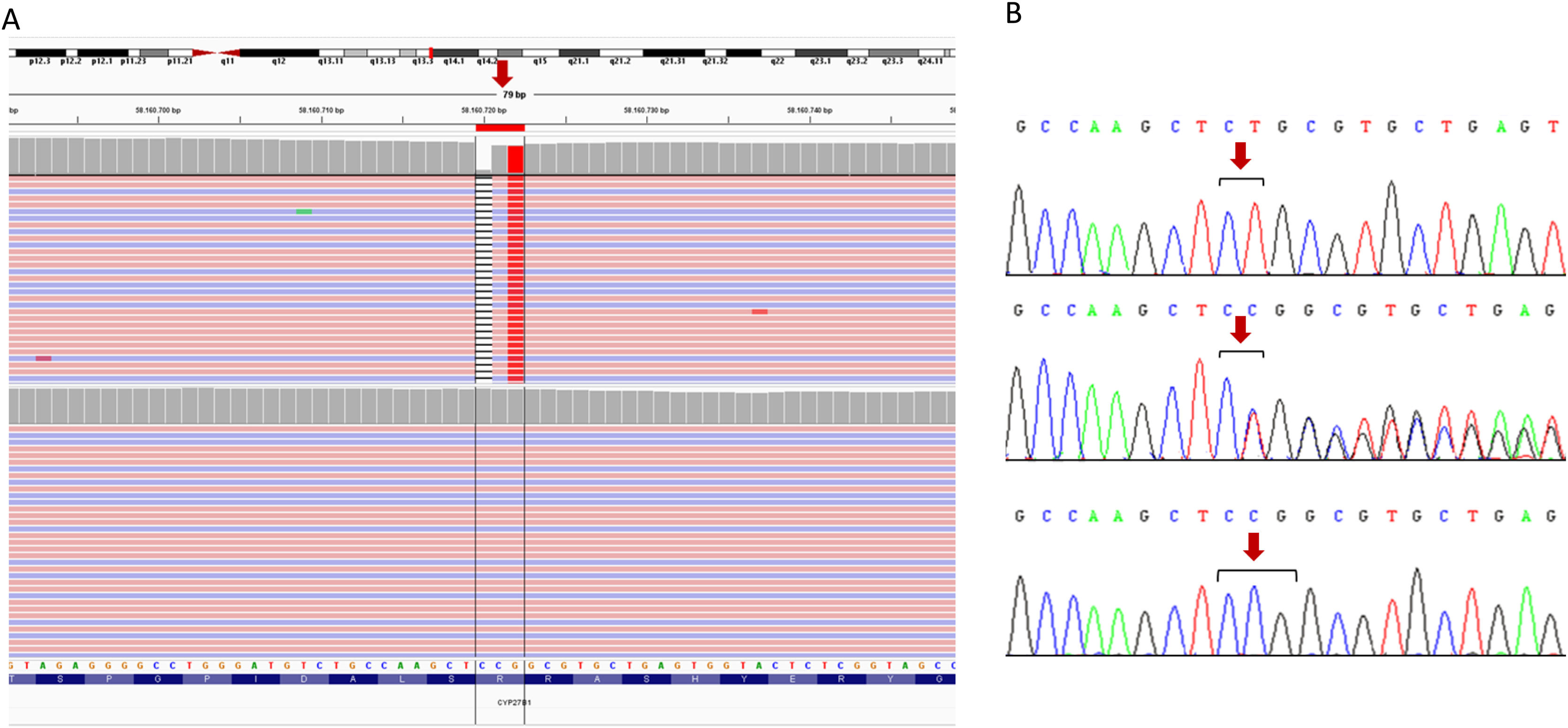
Familia 1Se identificó una variante en probable homocigosis en el exón 1 del gen CYP27B1 (NM_000785.4: c.103_104delinsA; p.(Ser36Alafs*42)) (fig. 1). El estudio mediante secuenciación Sanger, tanto en el caso 1 como en el 2 y en sus progenitores, confirmó la presencia de la variante familiar en homocigosis en ambas pacientes y en heterocigosis en sus progenitores. Descartaron consanguinidad.
 Identificación de la alteración en homocigosis en el exón 1 del gen CYP27B1 mediante secuenciación masiva (NGS). Los archivos obtenidos del alineamiento de las lecturas NGS paired-end (rosa, sentido; azul, antisentido) está cargado en el visor del genoma interactivo, IGV para su visualización. La secuencia del gen está en la orientación inversa en el cromosoma (hebra –1). Se indica la posición de la mutación mediante una flecha roja. La raya negra representa deleción, posteriormente se muestra el cambio por una timina (rojo) en homocigosis. El panel superior corresponde a la persona índice, mientras que el inferior a una persona control. B) Confirmación mediante secuenciación Sanger de la presencia de la variante c.103_104delinsA (nombrada en la hebra+1, según recomendaciones internacionales). El panel superior corresponde a la secuencia de la paciente índice, el intermedio a su padre y el inferior a una muestra control. Los resultados de la hermana y de la madre son idénticos a los mostrados en el panel superior y medio, respectivamente (datos no mostrados). Los nucleótidos alterados se muestran entre corchetes negros, la posición de la mutación se señala con una flecha roja.")
Resultados de los estudios genéticos realizados a la familia 1. A) Identificación de la alteración en homocigosis en el exón 1 del gen CYP27B1 mediante secuenciación masiva (NGS). Los archivos obtenidos del alineamiento de las lecturas NGS paired-end (rosa, sentido; azul, antisentido) está cargado en el visor del genoma interactivo, IGV para su visualización. La secuencia del gen está en la orientación inversa en el cromosoma (hebra –1). Se indica la posición de la mutación mediante una flecha roja. La raya negra representa deleción, posteriormente se muestra el cambio por una timina (rojo) en homocigosis. El panel superior corresponde a la persona índice, mientras que el inferior a una persona control. B) Confirmación mediante secuenciación Sanger de la presencia de la variante c.103_104delinsA (nombrada en la hebra+1, según recomendaciones internacionales). El panel superior corresponde a la secuencia de la paciente índice, el intermedio a su padre y el inferior a una muestra control. Los resultados de la hermana y de la madre son idénticos a los mostrados en el panel superior y medio, respectivamente (datos no mostrados). Los nucleótidos alterados se muestran entre corchetes negros, la posición de la mutación se señala con una flecha roja.
Se trata de una variante novel, no presente en bases de datos poblacionales como gnomAD, ExAC o 1000 Genomes, ni descrita en la literatura asociada a la enfermedad. El análisis bioinformático indica que la proteína truncada no es funcional, al eliminarse por completo los sitios de unión al ligando y por rotura del dominio con actividad vitamina 1α-hidroxilasa. Por todo ello, atendiendo a las guías de ACMG, esta variante se clasifica como probablemente patogénica (PVS1, PM2, PP4).
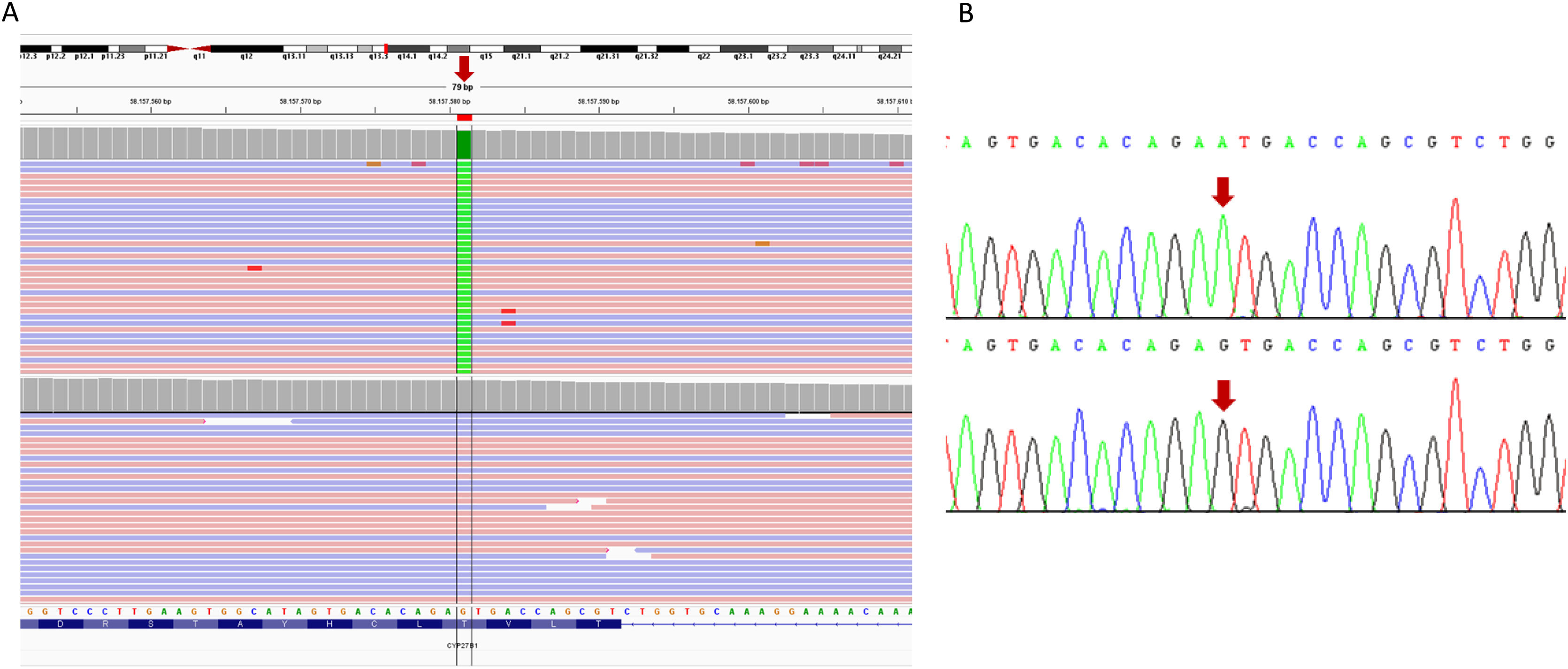
Familia 2Se identificó una variante en probable homocigosis en el exón 8 del gen CYP27B1 (NM_000785.4: c.1226C>T; p.(Thr409Ile)) (fig. 2). El algoritmo para el análisis de alteraciones en el número de copias (VarSeq CNV Caller, Golden Helix, Varseq 2.2.5) mostraba una ratio de ∼1 (computado con base en la cobertura media normalizada frente a muestras control), lo que descarta deleciones en este gen. La presencia de esta variante fue posteriormente confirmada por secuenciación Sanger en ambos pacientes. No se realizaron estudios de segregación al no disponer de muestra de progenitores.
 Visualización en el visor IGV de los resultados de la secuenciación NGS en los que aparece la sustitución de guanina por adenina (flecha roja), en homocigosis en el gen CYP27B1 en la paciente índice (panel superior), respecto a un control (panel inferior). La secuencia del gen está en la orientación inversa en el cromosoma. B) Validación, mediante secuenciación Sanger, de la presencia de la variante patogénica c.1226C>T (nombrada en la hebra+1, según recomendaciones internacionales). El panel superior corresponde a la secuencia del paciente índice y el inferior a una muestra control. La flecha indica la posición del nucleótido sustituido.")
Resultados de los estudios de secuenciación realizados a la familia 2. A) Visualización en el visor IGV de los resultados de la secuenciación NGS en los que aparece la sustitución de guanina por adenina (flecha roja), en homocigosis en el gen CYP27B1 en la paciente índice (panel superior), respecto a un control (panel inferior). La secuencia del gen está en la orientación inversa en el cromosoma. B) Validación, mediante secuenciación Sanger, de la presencia de la variante patogénica c.1226C>T (nombrada en la hebra+1, según recomendaciones internacionales). El panel superior corresponde a la secuencia del paciente índice y el inferior a una muestra control. La flecha indica la posición del nucleótido sustituido.
Se trata de una variante ya descrita como patogénica en varias familias independientes4.
Reevaluación clínicaFamilia 1La paciente 1, al ser informada del resultado genético, reveló que en la adolescencia presentaba piernas arqueadas, hipocalcemia y PTH elevada. En la actualidad, aqueja dolores óseos, especialmente en pies y rodillas. La densitometría ósea muestra baja densidad ósea en la columna lumbar y extremidad proximal del fémur. Se ha modificado la cantidad y periodicidad del tratamiento con calcitriol a 0,5μg/día para normalizar los niveles de PTH, 25(OH)D3 y 1,25(OH)2D3 y el dolor óseo se está tratando con tapentadol.
Su hermana menciona que en la adolescencia sufrió 2episodios de tetania, que presentaba dientes displásicos y piernas arqueadas. Además, en varias analíticas se detectó hipocalcemia, hiperfosfatemia, altos niveles de fosfatasa alcalina, de PTH y déficit de vitamina D. Cuando se le pautó tratamiento con calcitriol no volvió a presentar tetanias y se normalizaron los valores de vitamina D, tanto de 25(OH)D3 como de 1,25(OH)2D3.
Familia 2Al paciente 3, tras el resultado genético, se le modificó el tratamiento de calcidiol a calcitriol (0,25μg/día), manteniendo los suplementos cálcicos. La revisión de la historia clínica mostró que a los 10 años había sufrido convulsiones, clasificadas como febriles y, a los 14 años, episodios de crisis de hipertonía asociados a hipocalcemia generalizada. Un estudio de la serie ósea a los 23 años revelaba osteoesclerosis difusa de la columna y fémures con incurvación en forma de paréntesis. En la actualidad, reporta osteopenia en las rodillas.
A su hermana también se le modificó el tratamiento de calcidiol a calcitriol (0,25μg/día) y se le insistió en la necesidad de tomar suplementos cálcicos de manera continuada, sin éxito. A los 46 años aquejaba dolor coxofemoral bilateral e intermitente sin alteración detectable en estudio de densitometría ósea.
DiscusiónEl raquitismo dependiente de vitamina D (VDDR1) es una enfermedad con herencia autosómica recesiva que se caracteriza por la deficiencia de actividad 1α-hidroxilasa5. Las variantes patogénicas en el gen CYP27B1 están asociadas con VDDR1A (OMIM #264700), caracterizada por la presencia de hipotonía, retraso en el crecimiento y convulsiones o tetania. Asimismo, puede conllevar hipoplasia del esmalte dental durante la infancia6 y hallazgos de raquitismo de leves a moderados en las radiografías. Bioquímicamente cursa con concentraciones séricas bajas de calcio y fósforo junto con un aumento de los niveles de PTH y de fosfatasa alcalina. Asocian concentraciones normales o elevadas de 25(OH)D3 con niveles bajos de 1,25(OH)2D35.
En el presente trabajo, hemos identificado 2variantes en homocigosis en el gen CYP27B1. Ambas implican un defecto de la actividad enzimática de la 1α-hidroxilasa, en 2parejas de hermanos adultos, con variabilidad clínica intra- e interfamiliar, que coinciden con la dudosa correlación fenotipo-genotipo en el VDDR1A4, para la que se propone que factores como el tratamiento precoz con dosis variables de calcio pueden mitigar la clínica4.
Por otra parte, la rPTH es uno de los criterios mayores para el diagnóstico de iPPSD2. Sin embargo, los niveles elevados de PTH no siempre se deben a una resistencia, sino que pueden ser consecuencia de un mecanismo compensatorio activado por la deficiencia de vitamina D, dado que, cuando un paciente comienza a tener niveles insuficientes o deficientes de vitamina D, se produce un aumento compensatorio de la PTH hasta que los valores séricos de 1,25(OH)2D3 se normalizan7. No obstante, en pacientes con alteraciones en el metabolismo de la vitamina D, la concentración sérica de 1,25(OH)2D3 no se normaliza y los valores de PTH permanecen elevados7. Por este motivo se insiste en que la rPTH se define como la asociación de hipocalcemia, hiperfosfatemia y valores elevados de PTH en ausencia de déficit de vitamina D1,2.
Los pacientes descritos en este trabajo tenían sospecha clínica de PHP/iPPSD porque, de manera similar al VDDR1A, presentaban braquidactilia, hipocalcemia y valores de PTH elevados, y los niveles de vitamina D fueron el analito diferenciador entre ambas enfermedades1,2. Además, en los pacientes con VDDR1A la deficiencia de vitamina D suele aparecer tras el nacimiento y, normalmente, las manifestaciones clínicas se detectan durante los primeros años de vida5. Sin embargo, la falta grave de vitamina D en adultos es menos común y suele conllevar manifestaciones clínicas poco específicas relacionadas con osteomalacia, dolores óseos difusos y debilidad muscular6. Debido a que existen recomendaciones acerca de que no se solicite de manera rutinaria la determinación de los niveles de vitamina D8 y que las guías sobre los valores óptimos de vitamina D para el correcto funcionamiento de nuestro organismo se han consensuado de forma reciente9, el diagnóstico diferencial entre VDDR1A e iPPSD resulta complicado10. Sin embargo, queda demostrado que, en casos particulares, la determinación bioquímica de los niveles séricos tanto de 1,25(OH)2D3 como de 25(OH)D3 permiten orientar y realizar un diagnóstico adecuado.
Responsabilidades éticasLos estudios han sido revisados y aprobados por el Comité Ético de Investigación Clínica de Euskadi (CEI-E).
FinanciaciónEl presente trabajo ha sido financiado con el apoyo parcial del Instituto de Salud Carlos III del Ministerio de Economía y Competitividad (España), cofinanciado por el Fondo Europeo de Desarrollo Regional (subvención número PI20/00950) y el Departamento de Salud del Gobierno Vasco (subvención número GV2021/111056).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
