




La enfermedad del hígado graso no alcohólico (EHGNA) se ha convertido en el trastorno hepático más común en los países desarrollados, que abarca condiciones patológicas que van desde la esteatosis simple a la esteatohepatitis no alcohólica, cirrosis y hepatocarcinoma. A menudo la patogenia de la EHGNA ha sido interpretada por la hipótesis del «doble impacto», donde tras la acumulación de lípidos hepáticos tendría lugar la aparición de mediadores proinflamatorios que inducirían inflamación, lesión hepatocelular y fibrosis. Actualmente, el modelo propuesto sugiere que la constante exposición de los hepatocitos a ácidos grasos libres y sus metabolitos, agentes potencialmente lipotóxicos, estarían contribuyendo al desarrollo de EHGNA y resistencia hepática a la insulina; sugiriendo así un papel primordial para las lipasas metabólicas intracelulares en este proceso.
Non-alcoholic fatty liver disease (NAFLD) has become the most common liver disease in developed countries, covering a spectrum of pathological conditions ranging from single steatosis to non-alcoholic steatohepatitis, cirrhosis and hepatocellular carcinoma. Its pathogenesis has been often interpreted by the “double-hit” hypothesis, where the lipid accumulation in the liver is followed by proinflammatory mediators inducing inflammation, hepatocellular injury and fibrosis. Nowadays, a more complex model suggests that free fatty acids and their metabolites could be the true lipotoxic agents that contribute to the development of NAFLD and hepatic insulin resistance, suggesting a central role for metabolic lipases in that process.
La enfermedad del hígado graso no alcohólica (EHGNA) se ha convertido en el trastorno hepático más común en los países desarrollados, afectando aproximadamente a un 30% de adultos y a un 10% de niños1,2. Esta enfermedad abarca un espectro anatomopatológico de condiciones que van desde la simple acumulación de triglicéridos (TG) hepáticos (esteatosis simple) a la esteatosis con inflamación (esteatohepatitis), cirrosis e incluso hepatocarcinoma2,3. Aproximadamente el 20% de los pacientes con esteatohepatitis progresan hacia cirrosis e insuficiencia hepática4,5. De hecho, la esteatohepatitis asociada a cirrosis es actualmente la tercera causa más frecuente de trasplante hepático en los Estados Unidos6.
La EGHNA es considerada como la manifestación hepática del síndrome metabólico (SM) y se encuentra fuertemente asociada a la diabetes mellitus tipo 2 (DM2) y la obesidad. Tanto la obesidad como la DM2 son consecuencias del estilo de vida moderno, que se caracteriza por el aumento de la ingesta de ácidos grasos (AG) saturados, trans-insaturados y fructosa en la dieta, así como el sedentarismo7.
Aunque los mecanismos implicados en la patogénesis y progresión de la EHGNA no son del todo conocidos3, la resistencia a la insulina (RI) en el músculo, tejido adiposo y en el hígado parece desempeñar un papel central8,9. El deterioro en la señalización de la insulina en el tejido adiposo provoca un aumento de la lipólisis, generando así un flujo de AG hacia el hígado que promueven la RI hepática, con el consiguiente aumento de la lipogénesis de novo y acumulación de TG en este órgano10,11. Aproximadamente el 60% de los AG que participan en el cúmulo de TG hepáticos en la EHGNA proceden de la lipólisis del tejido adiposo, el 15% directamente de la dieta (ingesta excesiva de grasa e hidratos de carbono) y el 25% restante proceden del incremento de la ratio de la lipogénesis de novo controlada por factores de transcripción como SREBP1c (proteína de unión al elemento regulador de esterol), ChREBP (proteína de unión al elemento de respuesta a hidratos de carbono), LxRα (receptor X hepático) y PPARγ (receptor gamma activado por el factor proliferador de perixosomas). Muchos son los estudios que han demostrado el incremento de la expresión hepática de genes involucrados en la lipogénesis de novo en pacientes con EHGNA12–16. En este sentido, nuestro grupo, al estudiar la expresión de genes involucrados en el metabolismo lípidico hepático en mujeres obesas mórbidas con EGHNA, observó que la expresión hepática de FAS (sintasa de ácidos grasos), importante enzima lipogénica bajo el control de LxRα17, estaba significativamente aumentada en mujeres con esteatosis/esteatohepatitis no alcohólica, respecto a aquellas con histología hepática normal18.
Recientemente se ha observado que la constante exposición de los hepatocitos a metabolitos lipídicos potencialmente tóxicos, tales como AG, ácido fosfatídico, ácido lisofosfatídico, ceramidas y diacilgliceroles (DAG), pueden dar lugar a efectos «lipotóxicos»3, caracterizados por estrés en el retículo endoplásmico, inflamación, apoptosis, necrosis, ballooning y formación de cuerpos de Mallory-Denk, características histopatológicas propias de la esteatohepatitis19. Estas observaciones refuerzan la hipótesis de que los metabolitos derivados de los TG serían los verdaderos agentes tóxicos20,21, y que la hidrólisis de los TG hepáticos a través de lipasas metabólicas estarían contribuyendo al desarrollo de la EHGNA19.
Dado que actualmente no existen terapias efectivas para la EHGNA, a excepción de la pérdida de peso, los esfuerzos en la investigación actual se centran en la comprensión de la patogenia de esta enfermedad, con la finalidad de identificar nuevas dianas terapéuticas. En este sentido, esta revisión pretende actualizar los conocimientos sobre la contribución de las lipasas metabólicas hepáticas y mediadores lipotóxicos en el desarrollo de la esteatosis y esteatohepatitis no alcohólica.
Rol de las lipasas metabólicas en la patogénesis y progresión de la enfermedad del hígado graso no alcohólicoDebido a que la EHGNA se caracteriza principalmente por la acumulación hepática de lípidos22, las lipasas metabólicas se han involucrado en la patogénesis y progresión de la enfermedad y se encuentran actualmente en el centro de interés.
Los AG liberados del tejido adiposo son transportados hacia el hígado y captados mediante transportadores celulares específicos como clúster de diferenciación 36 (CD36) y proteína transportadora de ácidos grasos (FATP) o por difusión pasiva23. Una vez dentro de los hepatocitos son esterificados y almacenados en forma de TG. Estudios en ratones modificados genéticamente que sobreexpresan diacilglicerol aciltransferasa 2 en el hígado, enzima que cataliza el último paso en la formación de TG, han objetivado un aumento de la acumulación hepática de TG sin inflamación o RI hepática. Sin embargo, la silenciación de la expresión de diacilglicerol aciltransferasa 2 previene el cúmulo hepático de TG, pero causa daño lipotóxico debido al exceso de AG libres19. Estos hallazgos sugieren que los TG podrían estar protegiendo contra la lesión hepática mediada por lípidos. Sin embargo, las propiedades antilipotóxicas de los TG se ven limitadas debido a que las lipasas metabólicas pueden actuar sobre ellos liberando AG libres, generando así una nueva fuente de intermediarios lipotóxicos19.
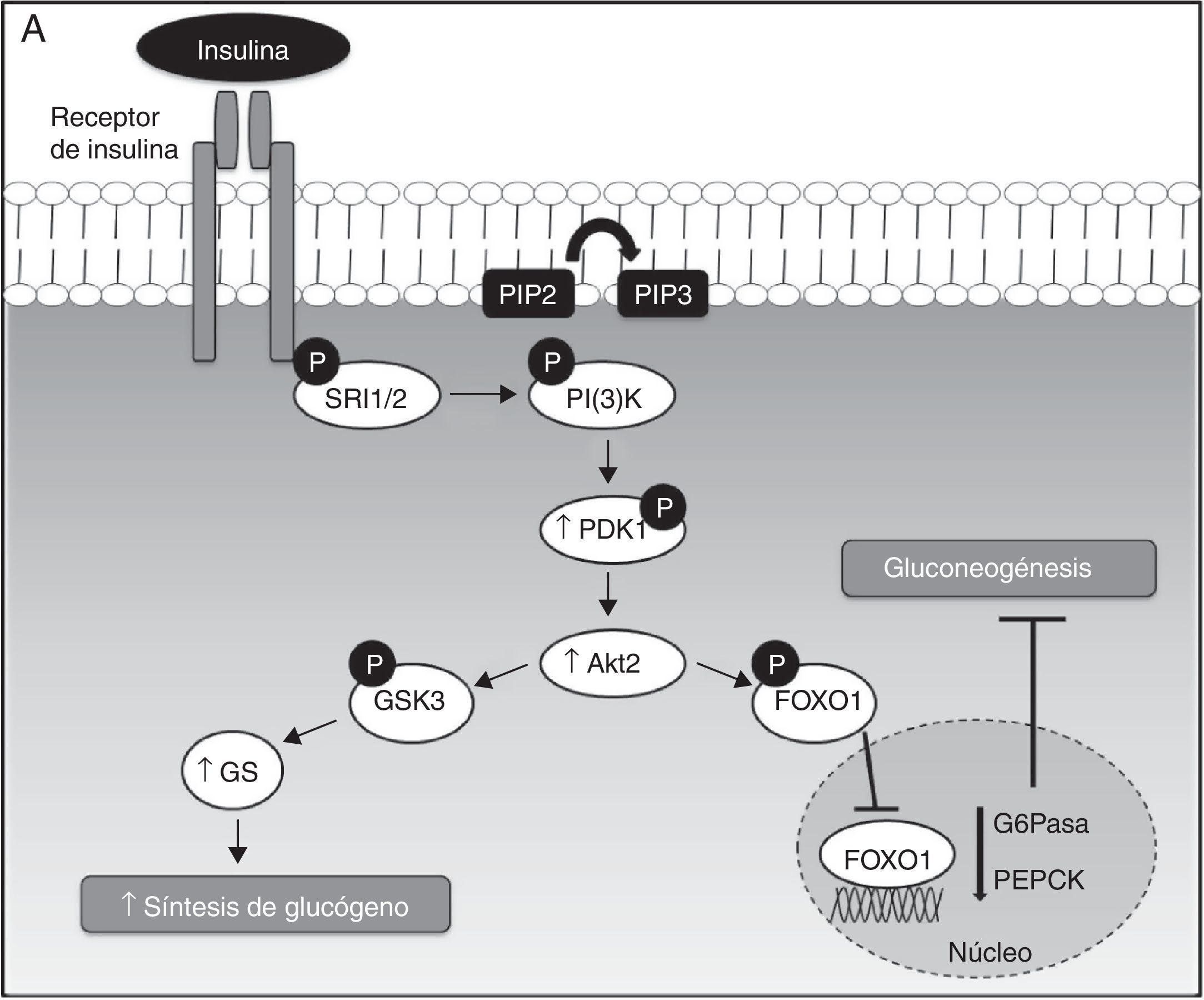
Diacilgliceroles, resistencia a la insulina hepática y enfermedad del hígado graso no alcohólicoLa acción de la insulina en las células hepáticas requiere un conjunto de señales intracelulares coordinadas, que se basan sobre todo en procesos de fosforilación y desfosforilación. Durante este proceso, la insulina se une a su receptor y activa la vía de la PI(3)K y Akt2, suprimiendo la producción hepática de la glucosa mediante 2 mecanismos principales: primero, la disminución de la expresión de las enzimas gluconeogénicas mediante la fosforilación y la exclusión nuclear del factor de transcripción FOXO1, y en segundo lugar, el aumento de la actividad glucógeno sintasa (GS), mediante la fosforilación e inactivación de la cinasa glucógeno sintasa 3 (GSK3)24–26 (fig. 1A).
, el cual promueve la fosforilación del sustrato del receptor de la insulina (SRI), siendo el SRI2 el más importante a nivel hepático. La fosforilación de SRI2 genera sitios de unión para la quinasa fosfatidilinositol 3 —PI(3)K—. La unión de PI(3)K a SRI2 convierte el lípido de membrana fosfatidilinositol 4,5-bifosfato (PIP2) en fosfatidilinositol 3,4,5-trifosfato (PIP3) que, a su vez, recluta Akt2. Bajo condiciones de estímulo de insulina, la proteína cinasa D (PDK1) es fosforilada y activa Akt2, que parece suprimir la producción hepática de glucosa mediante 2 mecanismos principales: primero, la disminución de la expresión de las enzimas gluconeogénicas mediante la fosforilación y la exclusión nuclear de la proteína FOXO1, que inhibe la activación de la expresión de proteínas gluconeogénicas como la glucosa-6 fosfatasa (G6Pasa) y la fosfoenolpiruvato carboxiquinasa (PEPCK), dando lugar a la supresión de la gluconeogénesis hepática; y en segundo lugar, el aumento de la actividad de la glucógeno sintasa (GS) mediante la fosforilación e inactivación de la quinasa glucógeno sintasa 3 (GSK3). En su forma inactiva (fosforilada), GSK3 no cataliza la fosforilación e inactivación de la GS, permitiendo así la síntesis de glucógeno hepática.B. Mecanismos moleculares de DAG-PKC¿ que median la resistencia a la insulina hepática. La acumulación hepática de DAG permite la activación y translocación de PKC¿ hacia la membrana plasmática, lo que provoca la inhibición del receptor quinasa de la insulina y su señalización intracelular.")
, el cual promueve la fosforilación del sustrato del receptor de la insulina (SRI), siendo el SRI2 el más importante a nivel hepático. La fosforilación de SRI2 genera sitios de unión para la quinasa fosfatidilinositol 3 —PI(3)K—. La unión de PI(3)K a SRI2 convierte el lípido de membrana fosfatidilinositol 4,5-bifosfato (PIP2) en fosfatidilinositol 3,4,5-trifosfato (PIP3) que, a su vez, recluta Akt2. Bajo condiciones de estímulo de insulina, la proteína cinasa D (PDK1) es fosforilada y activa Akt2, que parece suprimir la producción hepática de glucosa mediante 2 mecanismos principales: primero, la disminución de la expresión de las enzimas gluconeogénicas mediante la fosforilación y la exclusión nuclear de la proteína FOXO1, que inhibe la activación de la expresión de proteínas gluconeogénicas como la glucosa-6 fosfatasa (G6Pasa) y la fosfoenolpiruvato carboxiquinasa (PEPCK), dando lugar a la supresión de la gluconeogénesis hepática; y en segundo lugar, el aumento de la actividad de la glucógeno sintasa (GS) mediante la fosforilación e inactivación de la quinasa glucógeno sintasa 3 (GSK3). En su forma inactiva (fosforilada), GSK3 no cataliza la fosforilación e inactivación de la GS, permitiendo así la síntesis de glucógeno hepática.B. Mecanismos moleculares de DAG-PKC¿ que median la resistencia a la insulina hepática. La acumulación hepática de DAG permite la activación y translocación de PKC¿ hacia la membrana plasmática, lo que provoca la inhibición del receptor quinasa de la insulina y su señalización intracelular.")
A. Señalización hepática de la insulina. La insulina, a su llegada al hepatocito, se une y activa el receptor tirosina cinasa de la insulina (RTKI), el cual promueve la fosforilación del sustrato del receptor de la insulina (SRI), siendo el SRI2 el más importante a nivel hepático. La fosforilación de SRI2 genera sitios de unión para la quinasa fosfatidilinositol 3 —PI(3)K—. La unión de PI(3)K a SRI2 convierte el lípido de membrana fosfatidilinositol 4,5-bifosfato (PIP2) en fosfatidilinositol 3,4,5-trifosfato (PIP3) que, a su vez, recluta Akt2. Bajo condiciones de estímulo de insulina, la proteína cinasa D (PDK1) es fosforilada y activa Akt2, que parece suprimir la producción hepática de glucosa mediante 2 mecanismos principales: primero, la disminución de la expresión de las enzimas gluconeogénicas mediante la fosforilación y la exclusión nuclear de la proteína FOXO1, que inhibe la activación de la expresión de proteínas gluconeogénicas como la glucosa-6 fosfatasa (G6Pasa) y la fosfoenolpiruvato carboxiquinasa (PEPCK), dando lugar a la supresión de la gluconeogénesis hepática; y en segundo lugar, el aumento de la actividad de la glucógeno sintasa (GS) mediante la fosforilación e inactivación de la quinasa glucógeno sintasa 3 (GSK3). En su forma inactiva (fosforilada), GSK3 no cataliza la fosforilación e inactivación de la GS, permitiendo así la síntesis de glucógeno hepática.B. Mecanismos moleculares de DAG-PKC¿ que median la resistencia a la insulina hepática. La acumulación hepática de DAG permite la activación y translocación de PKC¿ hacia la membrana plasmática, lo que provoca la inhibición del receptor quinasa de la insulina y su señalización intracelular.
El desarrollo de la EHGNA se encuentra fuertemente asociado a la resistencia a la insulina hepática. Esta relación ha sido demostrada en ratas sometidas a una dieta rica en grasa durante 3 días, las cuales desarrollaron esteatosis y RI hepática, sin cambios en el peso corporal, adiposidad o RI en el músculo esquelético27. Curiosamente, en el hígado de estos animales se observó un aumento de DAG. La conexión entre la acumulación de DAG y RI hepática podría atribuirse a la activación de la proteína quinasa C¿ (PKC¿), altamente expresada en el hígado. Estos cambios se han asociado con reducciones en la fosforilación del receptor de la insulina y en la actividad Akt2. Por lo tanto, en este modelo, la actividad de la insulina para inducir la síntesis de glucógeno e inhibir la gluconeogénesis se ve disminuida debido a la activación de PKC¿ mediada por DAG, promoviendo la unión de PKC¿ en el dominio intracelular del receptor de la insulina (fig. 1B).
PKC¿ es un miembro de la familia PKC, compuesta por 3 grupos diferentes: convencional (α, βI, βII, y γ), novel (δ, ¿, η, y θ) y atípico (ζ y λ)28. PKC¿ es una isoforma novel con una afinidad mucho mayor por el DAG que las isoformas PKC convencionales29. El papel específico de PKC¿ en la RI hepática fue examinado en un estudio en el que se utilizaron oligonucleótidos antisentido (OAS), que actúan preferentemente en el hígado y el tejido adiposo30. Samuel et al. demostraron en ratas que la disminución de la expresión hepática de PKC¿ mediante OAS específicos las protegía del desarrollo de RI hepática inducida por lípidos, a pesar del aumento en el contenido de lípidos hepáticos31. Estos resultados fueron replicados en ratones knockout para el gen PKC¿, donde también se observó una protección frente al desarrollo de RI hepática inducida por una dieta rica en grasa32. Posteriormente, la interacción entre DAG, la activación de PKC¿ y la RI hepática ha sido demostrada en numerosos modelos de EHGNA asociada a resistencia hepática a la insulina33–44.
Estos modelos para la resistencia hepática a la insulina inducida por lípidos han sido translacionados a humanos. Kumashiro et al. evaluaron los posibles mecanismos implicados en la RI hepática en un grupo de pacientes con obesidad mórbida y EHGNA. En este caso, el contenido de DAG hepático y la activación de PKC¿ fueron los predictores más fuertes de RI hepática45. En cambio, no encontraron asociación entre la sensibilidad a la insulina y otros factores implicados en el desarrollo de RI hepática, tales como ceramidas, marcadores de estrés del retículo endoplásmico o concentraciones de citoquinas inflamatorias. Estos resultados fueron replicados en otro estudio en el que se demostró también el contenido de DAG hepático como el mejor predictor de la RI hepática en humanos obesos, mientras que no hubo asociación con el contenido de ceramidas o marcadores de inflamación hepáticos46. De hecho, se ha sugerido que la inflamación hepática es una consecuencia, y no una causa, de la resistencia a la insulina. Por lo tanto, aunque el exceso de ingesta de calorías conduce a la obesidad, solo aquellos que desarrollan esteatosis hepática desarrollarán RI. Estos resultados sostienen la hipótesis de que el paso clave en la patogénesis de la resistencia a la insulina hepática consiste en la acumulación de DAG, dando lugar a la activación de PKC¿.
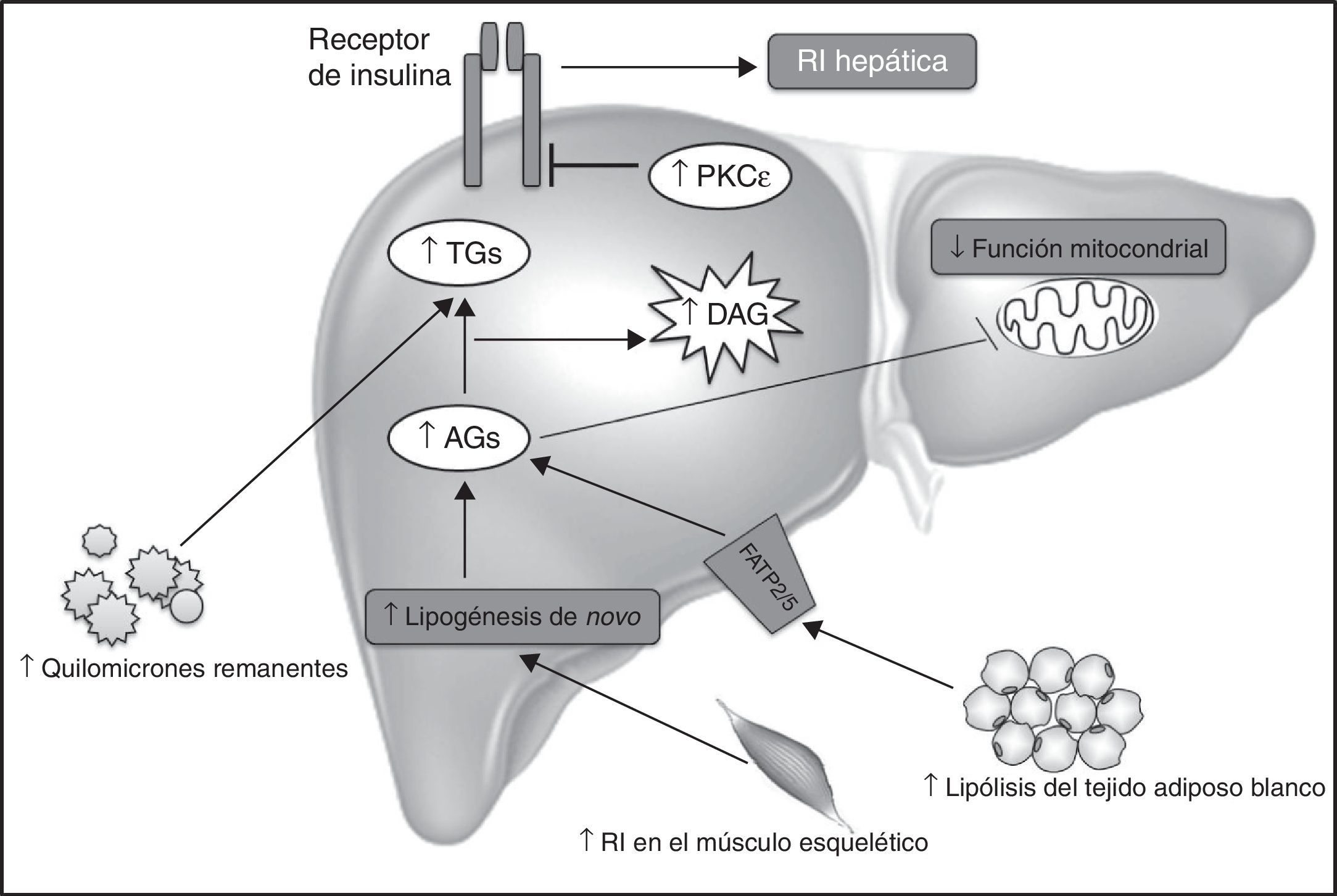
Mecanismos implicados en la acumulación hepática de diacilglicerolesLos DAG pueden acumularse en el hígado por diferentes motivos. En primer lugar, por un incremento del transporte de quilomicrones remanentes hacia el hígado. En segundo lugar, por un incremento de la liberación de AG por parte de los adipocitos. En tercer lugar, la hiperinsulinemia posprandial debido a la RI en el músculo esquelético puede derivar en un aumento de la lipogénesis de novo hepática, causando un aumento del contenido hepático de DAG. Finalmente, la disminución de la función mitocondrial puede también estar participando en la acumulación del contenido hepático de DAG47 (fig. 2). Cabe mencionar además que el sistema endocannabinoide parece tener un papel emergente en el desarrollo de RI hepática y acumulación de lípidos en el hígado.
, la tasa de oxidación mitocondrial de AG y la conversión de los DAG a triglicéridos (TG) durante la lipogénesis hepática. Que la ratio de la ingesta energética sea superior al gasto energético es una de las causas de la EHGNA y resistencia a la insulina (RI) hepática inducida por el mecanismo DAG-PKC¿. La predisposición de factores genéticos, como las variantes genéticas de APOC3 que provocan un aumento de las concentraciones plasmáticas APOC3, dan lugar a la supresión de la actividad de la lipoproteína lipasa, el aumento de quilomicrones remanentes posprandiales, y el aumento de la captación hepática de AG, contribuyendo al incremento del contenido hepático de DAG. Defectos en el almacenamiento lipídico en los adipocitos, tales como las lipodistrofias, así como alteraciones genéticas o adquiridas en la oxidación mitocondrial de AG, pueden contribuir a la acumulación hepática de DAG y consecuente desarrollo de EHGNA y RI hepática. Finalmente, los ácidos grasos liberados durante la lipólisis en los adipocitos entrarían en el hígado mediante transportadores específicos de AG (FATP2/5), aumentando el contenido hepático de DAG.")
Mecanismos implicados en la acumulación hepática de DAG.
El aumento en el contenido hepático de DAG es el resultado de un desequilibrio entre la captación/liberación de ácidos grasos (AG), la tasa de oxidación mitocondrial de AG y la conversión de los DAG a triglicéridos (TG) durante la lipogénesis hepática. Que la ratio de la ingesta energética sea superior al gasto energético es una de las causas de la EHGNA y resistencia a la insulina (RI) hepática inducida por el mecanismo DAG-PKC¿. La predisposición de factores genéticos, como las variantes genéticas de APOC3 que provocan un aumento de las concentraciones plasmáticas APOC3, dan lugar a la supresión de la actividad de la lipoproteína lipasa, el aumento de quilomicrones remanentes posprandiales, y el aumento de la captación hepática de AG, contribuyendo al incremento del contenido hepático de DAG. Defectos en el almacenamiento lipídico en los adipocitos, tales como las lipodistrofias, así como alteraciones genéticas o adquiridas en la oxidación mitocondrial de AG, pueden contribuir a la acumulación hepática de DAG y consecuente desarrollo de EHGNA y RI hepática. Finalmente, los ácidos grasos liberados durante la lipólisis en los adipocitos entrarían en el hígado mediante transportadores específicos de AG (FATP2/5), aumentando el contenido hepático de DAG.
Los endocannabinoides entran en las células hepáticas mediante transportadores específicos, tales como proteínas de unión de ácidos grasos (FABPs), FABP5 y FABP7, actuando48,49 sobre los receptores de canabinoides 1 (CB1) y 2 (CB2). Tanto los receptores de endocanabinoides como endocanabinoides específicos, como el 2-aciletanolamida (2-AE), se encuentran incrementados en el hígado de modelos de ratón con obesidad inducida por la dieta50,51. Además, en un estudio propio realizado en mujeres con obesidad mórbida y EHGNA, observamos que la expresión hepática de CB1 se encuentra significativamente incrementada en mujeres con esteatohepatitis comparadas con las que presentan únicamente esteatosis hepática52. Parece que la activación de CB1 podría activar la lipogénesis hepática mediante la inducción de estrés en el retículo endoplasmático, así como la activación de factores de transcripción (por ejemplo SREBP1c)53, proceso que estaría contribuyendo a la formación de DAG. Finalmente, el DAG acumulado podría ser transformado en 2-AE de nuevo, dando lugar a un bucle de retroalimentación positiva que estaría induciendo y agravando la esteatosis y la RI hepática54.
Aumento de la ingesta calóricaLa mayor causa de la HGNA en los países desarrollados se atribuye especialmente a un desequilibrio energético, donde la ingesta calórica excede al gasto calórico, conduciendo a un aumento de lípidos liberados hacia el hígado55–57.
En este sentido, Jonayvaz et al. demostraron que los ratones que seguían una dieta cetogénica rica en grasa desarrollaban esteatosis hepática severa y RI a pesar de manifestar un incremento de gasto energético y pérdida de peso. En este caso, el contenido de DAG hepático fue incrementado en un 350% debido a la activación de PKC¿, a la disminución de la fosfoliración de IRS2 inducida por insulina, y a la disminución de la supresión de la producción hepática de glucosa durante un clamp hiperinsulinémico-euglucémico.
En humanos diferentes estudios sugieren que la movilización regional de los TG circulantes y el transporte de ácidos grasos se encuentran alterados en los pacientes obesos que padecen EHGNA. La lipoproteína lipasa (LpL) hidroliza los TG circulantes, seguido por la absorción de estos en el tejido hepático a través de los transportadores celulares FATP y CD3658. La actividad LpL en el tejido adiposo en respuesta a la insulina parece estar disminuida en pacientes con obesidad59, mientras que la EHGNA se ha asociado con el incremento de la expresión hepática de LpL, FATP y CD3660–62. En general, parece que la sobreexpresión hepática de LpL63,64 o de CD36 provoca la acumulación hepática de lípidos, así como la resistencia a la insulina hepática65, mientras que la supresión hepática de FATP protege contra el desarrollo de esteatosis hepática e IR66.
En su conjunto, estos estudios sugieren que en la obesidad inducida por un aumento de la ingesta calórica los ácidos grasos son transportados desde el tejido adiposo hacia el hígado y el músculo esquelético, donde son reesterificados en DAG, induciendo así resistencia a la insulina en estos órganos.
Disminución de la función mitocondrialEn el hepatocito la β-oxidación mitocondrial es la principal ruta de oxidación de los ácidos grasos67. Las coenzimas reducidas flavín adenín dinucleótido y nicotinamida adenina dinucleótido, generadas en el propio proceso y en la oxidación del acetil-CoA mediante el ciclo de los ácidos tricarboxílicos, donan sus electrones a la cadena respiratoria, produciéndose adenosín trifosfato (ATP) por la fosforilación de la adenosín difosfato mediada por la ATP sintasa. Además, las mitocondrias son también la principal fuente de especies reactivas del oxígeno (ROS)68. Las ROS producidas en este y otros procesos son neutralizadas por sistemas enzimáticos, sobresaliendo la superóxido dismutasa, la catalasa y la glutatión peroxidasa, y por defensas celulares vitamínicas, principalmente la vitamina E y la C. En pacientes con EHGNA se ha descrito disfunción mitocondrial, anomalías ultraestructurales69,70, actividad reducida de los complejos de la cadena respiratoria68,71, fosforilación oxidativa deficiente, una menor capacidad para sintetizar ATP, un descenso en la concentración de ATP intracelular72 y daño en el ADN mitocondrial73.
Zhang et al.44 demostraron que la disminución de la función mitocondrial hepática puede ser un factor predisponente de EHGNA y RI hepática. Este estudio observó que los ratones Knockout para acil-CoA deshidrogenesa de cadena larga, los cuales tienen reducida la función mitocondrial en el hígado, son propensos a padecer esteatosis hepática asociada a un incremento del contenido de DAG, a la activación de PKC¿ y al desarrollo de RI hepática cuando estos son alimentados con una dieta rica en grasa. En humanos la asociación de la disminución de la oxidación hepática con EHGNA es menos clara, ya que existen estudios discrepantes. Algunos han demostrado una disminución de la oxidación74,75, mientras que otros han sugerido un incremento en el metabolismo mitocondrial hepático76.
Defectos en el almacenamiento lipídicoLas lipodistrofias son un grupo de síndromes de carácter congénito u adquirido cuya característica clínica principal es la pérdida parcial o completa del tejido adiposo. A nivel metabólico se caracterizan por una severa resistencia a la insulina, hipertrigliceridemia grave, bajos niveles circulantes de leptina, adiponectina y colesterol HDL, causado por la acumulación ectópica de grasa, lo que incluye el desarrollo de la EHGNA77. El 80% de los pacientes con lipodistrofia cumplen los criterios definitorios de SM78, sin embargo, presentan bajos niveles de hormonas derivadas de los adipocitos en comparación con pacientes con SM asociado a la obesidad73. Así, las lipodistrofias, situaciones en las que no existe expansión del tejido adiposo subcutáneo ni visceral, ofrecen una posibilidad única para evaluar de forma específica el papel del cúmulo hepático de grasa en el desarrollo de la EHGNA47.
Los ratones que expresan the dominant-negative protein A-ZIP/F1 carecen prácticamente de tejido adiposo blanco (ratones sin grasa) y desarrollan acumulación ectópica de grasa en el hígado y en el músculo esquelético, dando lugar a la aparición de una marcada RI periférica y hepática. Esta RI puede ser corregida mediante el trasplante de tejido adiposo blanco procedente de ratones sanos, lo que se traduce en una disminución de la cantidad grasa hepática acumulada y de la RI, tanto del tejido hepático como de tejidos periféricos79. Además, Shimomura et al., utilizando de nuevo un modelo murino de lipodistrofia, demostraron también su reversibilidad mediante la administración sistémica de leptina recombinante80.
En humanos se han estudiado también pacientes con lipodistrofia relacionada con la mutación en la perilipina-1, responsable de la inhibición de la triglicérido-lipasa intracelular. En estos pacientes, debido al aumento de la lipólisis, se observa una reducción del tejido graso subcutáneo y el desarrollo de EHGNA81. En los pacientes con lipodistrofia también se ha podido demostrar beneficio de la administración exógena de leptina recombinante82. Petersen et al., observaron que la administración exógena de leptina en pacientes lipodistróficos reducía significativamente el contenido lipídico hepático y muscular, debido principalmente a la disminución de la ingesta calórica, con la consiguiente mejora en la sensibilidad a la insulina tanto hepática como de los tejidos periféricos83. Recientemente, se ha comprobado que, en pacientes con diferentes grados de lipodistrofia, la terapia sustitutiva con leptina mejoraba el daño histológico del tejido hepático77.
En conjunto, los estudios realizados en modelos animales y en pacientes lipodistróficos demuestran una clara disociación entre la cantidad de tejido graso corporal global o la cuantía del tejido graso visceral, y el grado de RI hepática, sugiriendo que la distribución y acumulación específica del tejido graso al nivel del hígado y del músculo esquelético, no la cantidad grasa corporal total, es la que determina la RI hepática y muscular84.
Este último concepto estaría apoyado por el papel que el receptor PPARγ estaría desempeñando en el desarrollo de la esteatosis hepática. PPARγ está altamente expresado en el tejido adiposo, ejerciendo un papel clave en la captación de los ácidos grasos por parte de los adipocitos, así como en su diferenciación celular. Existen pacientes con ciertas mutaciones que tienen disminuida la actividad de PPARγ. Estos desarrollan síndrome metabólico y EHGNA, causado por el aumento del flujo lipídico hacia el hígado85. A pesar de que PPARγ está altamente expresado en el tejido adiposo, se expresa también a nivel hepático pero en menor grado. Ratones deficientes en la actividad hepática de PPARγ están protegidos contra el desarrollo de esteatosis hepática, sugiriendo que PPARγ desempeña un papel importante en la regulación del cúmulo de lípidos hepáticos86. Pacientes en tratamiento con tiazolidindionas, fármacos agonistas de PPARγ, presentan una disminución de la acumulación de lípidos a nivel hepático y del músculo esquelético, favoreciéndose el depósito lipídico en el tejido graso subcutáneo y el aumento de la sensibilidad hepática a la insulina87,88.
Así, los conocimientos aportados por los estudios de las lipodistrofias parecen apuntar a que la resistencia hepática a la insulina está determinada por el cúmulo específico de grasas a nivel hepático, y no por el grado de obesidad global, y en segundo lugar, resaltan la importancia del tejido adiposo y de su capacidad adaptativa para almacenar el exceso de grasas, hipertrofiándose, protegiendo al hígado de un cúmulo lipídico excesivo.
Resistencia a la insulina en el músculo esqueléticoEn este apartado se pretende dilucidar el papel de la RI muscular en la patogénesis de la EHGNA y la RI hepática.
Papel del exceso de ácidos grasos libres en la resistencia a la insulina muscularEl potencial de los ácidos grasos libres (AGL) para alterar el metabolismo glucídico muscular fue propuesto hace más de 50 años89, y desde entonces ha sido ampliamente investigado. Diferentes autores90–96 han confirmado que un incremento en la concentración plasmática de AGL altera la señalización de la insulina y causa insulinorresistencia muscular en individuos sanos. Además, se ha demostrado que una disminución de la concentración plasmática de AGL secundaria a acipimox, análogo del ácido nicotínico e inhibidor de la lipólisis del tejido adiposo, mejora rápidamente la sensibilidad a la insulina muscular97.
Así mismo, numerosos estudios han demostrado una fuerte relación entre los lípidos intramiocelulares y la RI muscular55,56,98. En adultos no diabéticos con normopeso el contenido en TG intramiocelular es un predictor de RI muscular mucho más potente que los ácidos grasos circulantes99, lo que sugiere que, en sujetos obesos, los lípidos intramiocelulares pueden estar desempeñando un papel causal en la RI muscular. De hecho, sujetos obesos insulinosensibles o insulinorresistentes se distinguen según el cúmulo lipídico muscular y hepático100.
Por otro lado, es importante señalar que la RI muscular correlaciona con una variedad de metabolitos lipídicos tóxicos, consecuencia de una oxidación incompleta de ácidos grasos, como acilcarnitinas, ceramidas y DAG96,101–104. En modelos murinos, cuando se aumenta los AG en plasma mediante la infusión de Liposyn®+heparina para activar la LpL, la RI muscular aparece a las 3h, a consecuencia del aumento de DAG y de la activación de PKC¿105. Sin embargo, no se observan cambios en el contenido de TG o ceramida muscular en ese momento, por lo que es importante diferenciar entre los distintos lípidos que participan en la patogénesis de la RI muscular. Los hallazgos de RI muscular mediada por DAG han sido confirmados en humanos por Itani et al.106. Además, en la patogénesis de RI muscular, es importante tener en cuenta que el músculo esquelético también es el objetivo de citoquinas proinflamatorias circulantes como factor de necrosis tumoral alfa e interleucina 6. Finalmente, macrófagos inflamatorios M1 activados procedentes del tejido adiposo pueden infiltrar el músculo esquelético y causar RI de una manera similar a lo que ocurre en el tejido adiposo107.
Insulinorresistencia en músculo esquelético en pacientes con esteatohepatitis no alcohólicaSegún la literatura un aumento moderado en la adiposidad y en la concentración plasmática de AGL puede causar lipotoxicidad muscular. Por tanto, no es de extrañar que, en pacientes obesos con EHGNA, la RI muscular esté completamente establecida108–110, e incluso en individuos no obesos con esteatohepatitis no alcohólica (EHNA) si presentan RI en el tejido adiposo111–113. En cambio, individuos obesos sin RI en el tejido adiposo se comportan desde el punto de vista metabólico como sujetos delgados, con casi normal insulinosensitividad muscular y habitualmente sin desarrollo de EHGNA108,111,114.
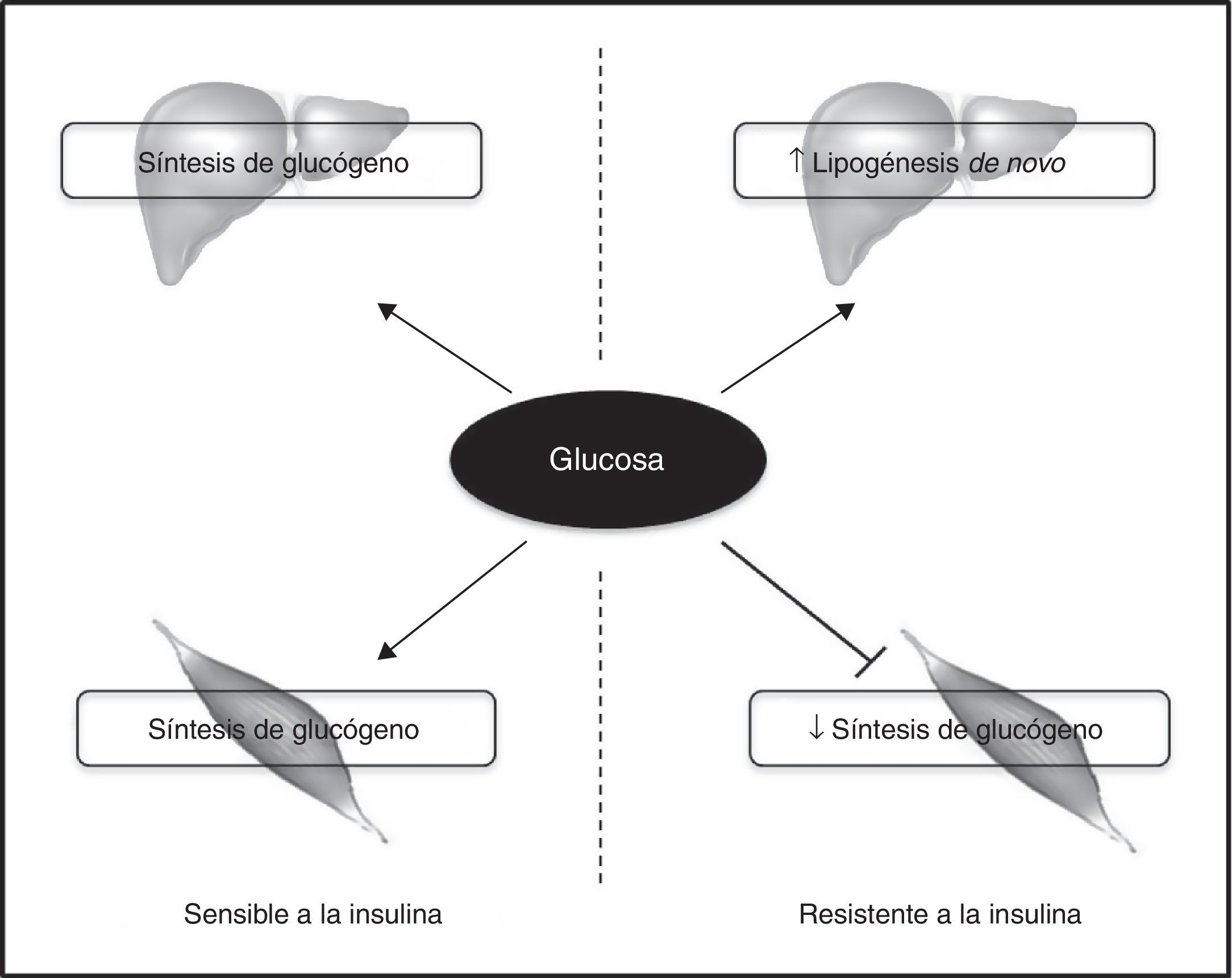
La RI primaria en el músculo esquelético puede conducir a una redistribución de los sustratos hacia el hígado, dando lugar a la aparición de esteatosis hepática y, posteriormente, RI hepática a través del cúmulo hepático de DAG con la activación de PKC¿. Esta hipótesis ha sido respaldada en estudios de modelos murinos que presentan una inactivación específica del gen del receptor de insulina (MIRKO)115. La RI muscular selectiva en estos ratones comporta una hiperinsulinemia compensatoria con redistribución de los sustratos hacia lugares ectópicos, tales como el hígado. Además, ratones que carecen del transportador de glucosa muscular (GLUT4) presentan una pérdida casi completa de la captación muscular de glucosa estimulada por la insulina, que se asocia a esteatosis hepática y RI116. Estos hallazgos se han corroborado también en humanos. Así, en individuos con RI muscular selectiva, observada en sujetos sanos, jóvenes, delgados o en individuos en el cuartil inferior de insulinosensibilidad global, los hidratos de carbono ingeridos se desvían de la síntesis de glucógeno muscular hacia la lipogénesis hepática, predisponiendo a estos individuos a RI hepática y aparición de EHGNA117 (fig. 3). Por otra parte, un estudio reciente ha demostrado que una sola sesión de ejercicio revierte los defectos en el transporte de la glucosa muscular estimulada por insulina, así como los defectos en la síntesis de glucógeno118. Esta mejoría se evidencia por una disminución de la lipogénesis hepática de novo y por la reducción de la síntesis hepática de TG neta después de una dieta rica en hidratos de carbono. Por lo tanto, se demuestra que la RI muscular puede ser una diana terapéutica para la prevención y el tratamiento de la EHGNA118. En este sentido, Haufe et al. demostraron que el efecto de la mejora de la condición física sobre la insulinosensibilidad en pacientes con sobrepeso/obesidad está mediada a través de una reducción en el contenido de grasa hepática119. Además, se ha objetivado que el ejercicio crónico sin reducción en el peso corporal o en la grasa corporal total también conduce a una reducción en el contenido de grasa hepática120.

La resistencia a la insulina en el músculo esquelético contribuye a la lipogénesis hepática.
En los sujetos insulinosensibles la insulina estimula la síntesis de glucógeno tanto en tejido muscular como hepático. Sin embargo, en aquellos que presentan resistencia a la insulina en el músculo esquelético, esta hormona no es capaz de promover la síntesis de glucógeno, redistribuyendo el sustrato hacia el hígado, lo que contribuye a un incremento de la lipogénesis de novo hepática. El aumento de la síntesis de lípidos en el hígado de pacientes con resistencia a la insulina en el músculo esquelético promueve el desarrollo de EHGNA.
En resumen, tanto estudios en ratones como en humanos apoyan el concepto de que la RI muscular selectiva, que es una de las alteraciones metabólicas más tempranas detectadas en jóvenes delgados descendientes de padres con diabetes tipo 2, puede ser un factor importante y temprano en la patogénesis de la EHGNA y la resistencia hepática a la insulina.
Factores genéticosEn los últimos años se han descrito variantes genéticas de distintos genes que hacen más suceptibles a los individuos portadores a desarrollar EHGNA y RI cuando se exponen a factores ambientales.
Apolipoproteína C3La apolipoproteína C3 (APOC3) es una proteína que forma parte de las lipoproteínas de muy baja densidad, que se encarga de inhibir la actividad lipoproteín lipasa con el fin de regular la distribución de lípidos en los diferentes órganos y tejidos del cuerpo.
Recientemente, Petersen et al. han demostrado que un grupo de indios asiáticos, varones, sanos y delgados, que presentan una de las 2 variantes genéticas (C-482T/T-455C) en el elemento de respuesta a la insulina del gen APOC3 se encuentran en un riesgo más elevado de desarrollar EHGNA y RI. Las concentraciones plasmáticas de APOC3, así como la hipertrigliceridemia posprandial de los individuos que poseen este polimorfismo, son aproximadamente un 30% más elevadas comparadas con los sujetos homozigotos para APOC3 (C-482/T-455)121. Además, después de ser sometidos a una infusión intravenosa de lípidos, estos sujetos presentaron una disminución de la hidrólisis de triglicéridos y un aumento de la hipertrigliceridemia posprandial y quilomicrones remanentes, debido a la exagerada inhibición de la actividad LpL, permitiendo así el desarrollo de EHGNA y RI. También se pudo observar una reversión de la esteatosis hepática y RI en estos sujetos después de una modesta pérdida de peso121. Sin embargo, cabe destacar que las variantes genéticas de APOC3 no son la causa directa del desarrollo de esteatosis hepática, pero suponen una condición de predisposición en aquellos individuos que son portadores, siendo más susceptibles a desarrollar EHGNA y RI cuando se exponen a factores ambientales tóxicos, tales como una dieta hipercalórica y rica en grasas. De acuerdo a esta hipótesis, Lee et al. han demostrado recientemente que ratones que sobreexpresan APOC3 no presentan esteatosis hepática o RI cuando son alimentados con dieta control. Sin embargo, cuando esta es remplazada por una dieta rica en grasas desarrollan esteatosis hepática severa con un incremento del contenido hepático de DAG, de la activación de PKC¿ y RI40.
AdiponutrinOtro grupo en riesgo de desarrollo de EHGNA y RI hepática son adultos y niños hispanos122. En esta población se ha visto que la sustitución de isoleucina por metionina en la posición 148 (I148M) de la enzima PNPLA3, conocida también como adiponutrin, codificada por el gen PNPLA3, se encuentra fuertemente asociada al contenido hepático de TG y desarrollo de EHGNA, incluyendo esteatohepatitis, cirrosis y hepatocarcinoma123–127. Sin embargo, a diferencia de las variantes del gen APOC3 descritas en la sección anterior, las variantes genéticas de PNPLA3 no se encuentran asociadas a RI, sugiriendo que la enfermedad de EHGNA puede disociarse de la RI hepática128–130.
PNPLA3 pertenece a la familia de proteínas que comparten un dominio evolutivo identificado por primera vez en la patatina, la principal proteína de los tubérculos de patata131. El genoma humano codifica 9 proteínas que contienen el dominio de la patatina, de los cuales PNPLA3 está estrechamente relacionada con PNPLA2, la mayor hidrolasa de TG del tejido adiposo132,133. PNPLA3 se expresa predominantemente en el hígado y en el tejido adiposo humano134, y se encuentra frecuentemente asociada a membranas y vacuolas lipídicas135. Esta proteína está altamente regulada en respuesta a estímulos nutricionales, tanto a nivel transcripcional como postranslacional. Su expresión se suprime en estado de ayunas, mientras que aumenta en respuesta a la presencia de glucosa e insulina, donde el factor de transcripción SREBP1c se encarga de estimular su transcripción136.
Se sugiere que PNPLA3 presenta tanto función lipasa como de transacilación, y que la mutación I148M causa la pérdida de la función lipasa del gen137, contribuyendo así al aumento del contenido de TG hepáticos. Yongcheng et al. demostraron que PNPLA3 cataliza la hidrólisis de los 3 mayores glicerolípidos (TG, DAG y MAG), con mayor preferencia por los ácidos grasos C18:1, y que la mutación I148M reduce la actividad hidrolasa contra los 3 glicerolípidos. Sin embargo, cabe destacar que, prácticamente, en la mayoría de estos estudios se han incluido individuos obesos como sujetos controles, los cuales presentan casi siempre algún grado de esteatosis hepática asociada a RI, haciendo difícil discernir si las variantes genéticas de PNPLA3 con EHGNA están participando o no en la RI hepática. Para identificar si la mutación confiere un riesgo independiente en el desarrollo de RI hepática, sería importante evaluar la sensibilidad a la insulina hepática en individuos delgados con EHGNA portadores de la mutación I148M136.
En concordancia a los resultados obtenidos en humanos, la sobreexpresión del gen mutante I148M de PNPLA3 en ratones se asoció a un aumento del tamaño de las vacuolas lipídicas y acumulación de TG hepáticos135,136. Sin embargo, la sobreexpresión de PNPLA3 no mutado no cambió el contenido de lípidos hepáticos en ratones135,136,138. Por otro lado, los ratones Knockout para PNPLA3 no desarrollaron esteatosis hepática ni trastornos en el metabolismo de la glucosa139,140.
Rol de la compartimentación intracelular de diacilgliceroles: disociación de la enfermedad del hígado graso no alcohólico y la resistencia a la insulina hepáticaEl modelo descrito anteriormente, que explica la relación entre DAG, PKC¿ y RI hepática, se centra principalmente en la incapacidad de la insulina para alterar el metabolismo de la de glucosa hepática. Sin embargo, la capacidad de la insulina para activar la lipogénesis parece estar intacta en la mayoría de los modelos de EHGNA. Por ello, aunque la EHGNA se encuentra fuertemente asociada a RI hepática y DM2, recientemente se ha descrito la «paradoja de la resistencia hepática a la insulina selectiva», en la que se propone la existencia de una disociación entre la EHGNA y la RI hepática141,142.
Experimentalmente es posible inducir resistencia a la insulina sin EHGNA, o inducir EHGNA sin resistencia a la insulina bajo ciertas condiciones. Por ejemplo, el bloqueo de la secreción hepática de lipoproteínas de muy baja densidad con una dieta deficiente en colina, o mediante la modificación genética de la maquinaria de exportación, aumenta las concentraciones de TG hepáticos pero no induce RI en ratones143,144. Por otro lado, los factores de transcripción ChREBP y SREBP1 han sido recientemente implicados, de forma independiente, en la disociación de EHGNA y RI hepática tanto en ratones como en humanos145,146. Para explicar esta paradoja y la RI hepática selectiva, Cantley et al. examinaron la localización subcelular de DAG en células hepáticas y observaron que el knockdown del gen CGI-58, activador de la lipasa de triglicéridos del tejido adiposo (ATGL) que participa en la hidrólisis de TG, promueve la acumulación de DAG en gotas lipídicas, evitando así la acumulación de DAG en la membrana celular y la translocación de PKC¿ hacia la membrana celular147. Estos resultados concuerdan con otros estudios realizados tanto en humanos45 como en modelos animales38,148,149, sugiriendo que la compartimentación de DAG en el hepatocito podría ser un factor importante en la patogénesis de la resistencia hepática a la insulina.
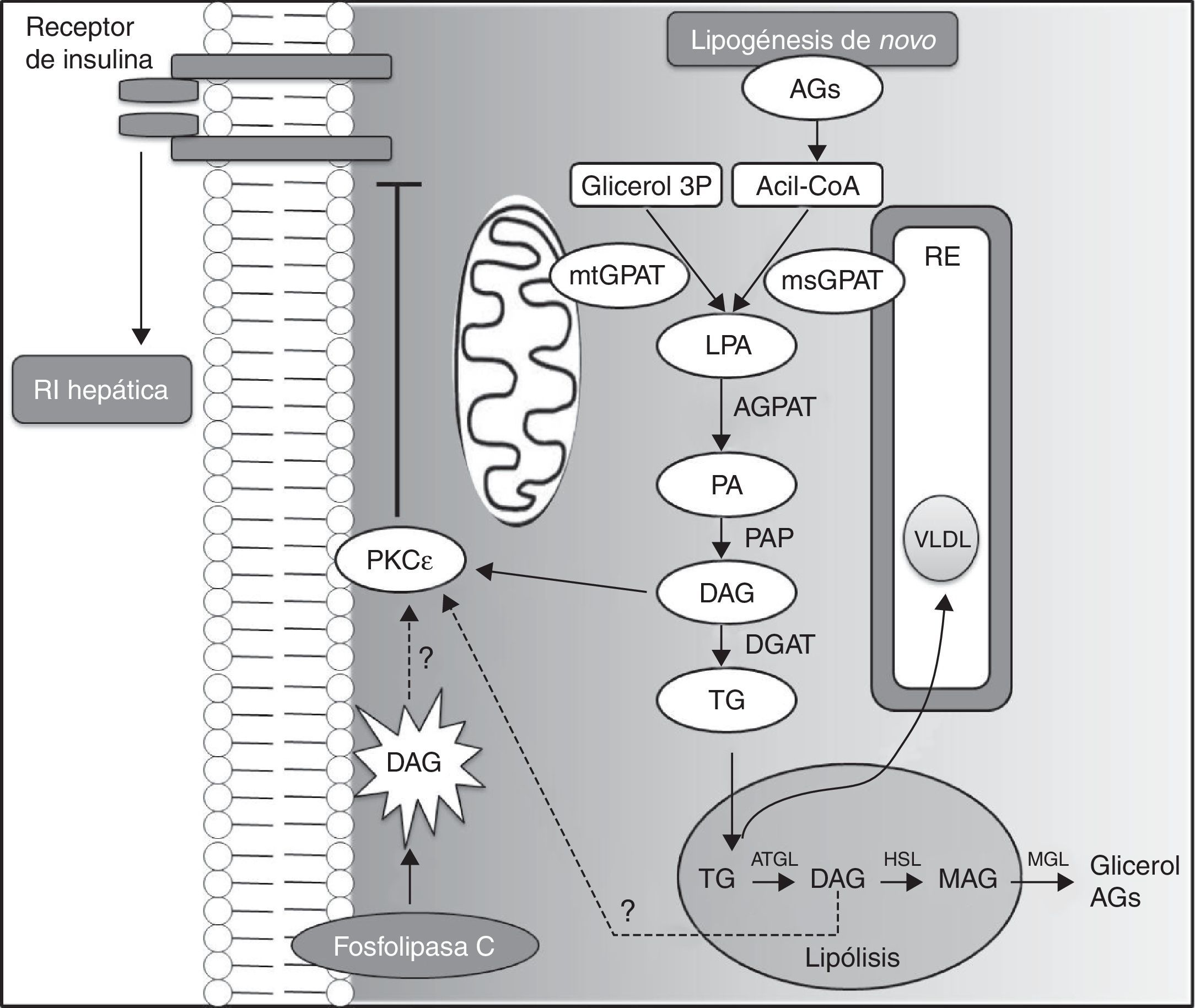
La acumulación hepática de DAG puede resultar de la vía glicerol 3-fosfato, que representa la ruta de la lipogénesis en la síntesis de TG y fosfolípidos. Por otro lado, los DAG intracelulares también pueden derivar de la hidrólisis de TG compartimentados en gotas lipídicas, mediada por la lipasa ATGL, así como de la activación de la fosfolipasa C, que libera DAG a partir de lípidos de membrana (fig. 4). De qué manera los DAG que derivan de estas 2 últimas vías pueden conducir a la activación PKC¿ y a la RI hepática no se ha determinado todavía. Sin embargo, los resultados obtenidos usando OAS específicos para CGI-58 indican claramente que los lípidos secuestrados en gotas lipídicas no promueven la activación PKC¿ y resistencia hepática a la insulina, y que la compartimentación de DAG y TG en compartimentos neutros, en los cuales se estaría evitando la activación de PKC¿, podrían explicar por qué el contenido hepático de DAG no siempre se correlaciona con la RI hepática en algunos modelos animales47.
 representa la ruta de la lipogénesis de novo en la síntesis de triglicéridos y fosfolípidos. La glicerol-fosfato-acil-transferasa (GPAT) cataliza la acetilación de glicerol 3P con acil-CoA para generar ácido lisofosfatídico (LPA), lo que parece ser el paso limitante en la síntesis de triglicéridos (TG). Posteriormente, las enzimas acil-glicerol-fosfato-acil-transferasa (AGPAT), fosfatasa del ácido fosfatídico (PAP) y diacilglicerol aciltransferasa (DGAT) catalizan la formación de ácido fosfatídico (PA), diacilglicerol (DAG) y TG, respectivamente. Los DAG activan la translocación de PKC¿ hacia la membrana plasmática, inhibiendo la activación del receptor de la insulina. Los ácidos LPA y PA que no han sido sintetizados en el retículo endoplasmático (RE) requieren la translocación a través del citosol para la síntesis de TG en el RE. En el hígado los TG pueden depositarse en vacuolas intracelulares o ser exportados en lipoproteínas de muy baja densidad (VLDL). En las vacuolas, durante la lipólisis, la conversión de TG a DAG es mediada por la lipasa de triglicéridos del tejido adiposo (ATGL). Los DAG pueden ser hidrolizados a monoacilglicerol (MAG) por la hormona sensible a la lipasa (HSL) y, posteriormente, a glicerol por la lipasa de monoglicéridos (MGL), que puede ser utilizado como sustrato para la gluconeogénesis. Estas reacciones, al liberar ácidos grasos, contribuyen a su acumulación hepática. Los DAG provenientes de lipólisis intravacuolar y de la vía de la fosfolipasa C, que libera DAG a partir de lípidos de membrana, podrían activar la PKC¿ y la resistencia hepática a la insulina (RI). Sin embargo, estos 2 últimos mecanismos no están del todo dilucidados.")
Vías metabólicas implicadas en la acumulación hepática de DAG.
La vía del glicerol-3-fosfato (glicerol 3P) representa la ruta de la lipogénesis de novo en la síntesis de triglicéridos y fosfolípidos. La glicerol-fosfato-acil-transferasa (GPAT) cataliza la acetilación de glicerol 3P con acil-CoA para generar ácido lisofosfatídico (LPA), lo que parece ser el paso limitante en la síntesis de triglicéridos (TG). Posteriormente, las enzimas acil-glicerol-fosfato-acil-transferasa (AGPAT), fosfatasa del ácido fosfatídico (PAP) y diacilglicerol aciltransferasa (DGAT) catalizan la formación de ácido fosfatídico (PA), diacilglicerol (DAG) y TG, respectivamente. Los DAG activan la translocación de PKC¿ hacia la membrana plasmática, inhibiendo la activación del receptor de la insulina. Los ácidos LPA y PA que no han sido sintetizados en el retículo endoplasmático (RE) requieren la translocación a través del citosol para la síntesis de TG en el RE. En el hígado los TG pueden depositarse en vacuolas intracelulares o ser exportados en lipoproteínas de muy baja densidad (VLDL). En las vacuolas, durante la lipólisis, la conversión de TG a DAG es mediada por la lipasa de triglicéridos del tejido adiposo (ATGL). Los DAG pueden ser hidrolizados a monoacilglicerol (MAG) por la hormona sensible a la lipasa (HSL) y, posteriormente, a glicerol por la lipasa de monoglicéridos (MGL), que puede ser utilizado como sustrato para la gluconeogénesis. Estas reacciones, al liberar ácidos grasos, contribuyen a su acumulación hepática. Los DAG provenientes de lipólisis intravacuolar y de la vía de la fosfolipasa C, que libera DAG a partir de lípidos de membrana, podrían activar la PKC¿ y la resistencia hepática a la insulina (RI). Sin embargo, estos 2 últimos mecanismos no están del todo dilucidados.
Las lipasas metabólicas son las enzimas responsables de la hidrólisis de TG tanto en tejido adiposo como hepático. La lipólisis en el tejido adiposo provoca un aumento del flujo de AG hacia el hígado, promoviendo así el desarrollo de esteatosis hepática150. Existe una creciente evidencia, no solo en modelos animales de EHGNA, sino también en humanos con EHGNA asociada a obesidad, a DM2 y a lipodistrofia, que la esteatosis hepática está fuertemente vinculada al desarrollo de RI hepática. Por lo tanto, si los lípidos hepáticos son mediadores importantes para la RI hepática, es de esperar que la reducción de la esteatosis hepática en pacientes con EHGNA y DM2 conlleve a la reducción de la RI hepática.
La EHGNA aparece cuando la ratio de lípidos suministrados hacia el hígado excede a la oxidación y a la exportación lipídica hepática. Por otro lado, un gran número de estudios, tanto en seres humanos como en modelos animales, han demostrado que la acumulación hepática de DAG permite la activación de PKC¿, dando lugar a la aparición de resistencia hepática a la insulina. Además, la compartimentación del DAG acumulado parece ser un factor crucial para el desarrollo de RI hepática, pudiendo explicar por qué algunos pacientes y modelos murinos con EHGNA no desarrollan RI hepática. Así, el mecanismo DAG-PKC¿ parece desempeñar un papel primordial en el desarrollo de la EHGNA asociada a la RI hepática. Por todo ello, para el tratamiento de la EHGNA y la DM2 resulta de gran interés todas aquellas terapias que van dirigidas a reducir la liberación de ácidos grasos hacia el hígado, a suprimir la producción de DAG o a aumentar la oxidación mitocondrial de los AG151.
Aparte de la pérdida de peso, no existen actualmente terapias efectivas para la EHGNA. Por dicho motivo, la investigación actual se centra en la comprensión de la enfermedad subyacente a la esteatosis hepática, con la finalidad de identificar nuevas dianas terapéuticas. En este sentido, se ha sugerido que nuevas terapias dirigidas a prevenir la acumulación hepática de DAG y la activación de PKC¿ podrían ser efectivas84. Por otro lado, pequeños péptidos dirigidos a revertir la función enzimática correcta de las variantes genéticas de PNPLA3 asociadas a la progresión de la EHGNA y la esteatohepatitis no alcohólica podrían proporcionar un enfoque innovador para el tratamiento de la EHGNA.
FinanciaciónEste trabajo ha recibido financiación del Fondo de Investigación Sanitaria (expediente n.° PI13/00468 de Teresa Auguet).
AutoríaAlba Berlanga y Esther Guiu-Jurado han contribuido por igual en este trabajo. Todos los autores han contribuido en la redacción del artículo. La versión final de este manuscrito está aprobada por todos los autores.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.