




Los estudios de asociación basados en genes candidato llevados a cabo durante décadas han servido para visualizar la influencia del componente genético en enfermedades complejas como la arteriosclerosis y también para evidenciar la interacción entre diferentes genes y, especialmente, la de estos con factores ambientales. Incluso con el conocimiento acumulado, aún queda camino por recorrer para descifrar la predisposición individual a la enfermedad y, si tenemos en cuenta la gran influencia que los factores ambientales juegan en el desarrollo y la progresión de la arteriosclerosis, la epigenética se nos presenta como un elemento clave para ampliar nuestro conocimiento de la predisposición individual a la arteriosclerosis.
La epigenética se puede describir como la disciplina que estudia los mecanismos de regulación transcripcional independientes de la secuencia del ADN, mayoritariamente inducidos por factores ambientales.
Esta revisión pretende describir qué es la epigenética y de qué manera los mecanismos epigenéticos participan en la arteriosclerosis.
The association studies based on candidate genes carried on for decades have helped in visualizing the influence of the genetic component in complex diseases such as atherosclerosis, also showing the interaction between different genes and environmental factors. Even with all the knowledge accumulated, there is still some way to go to decipher the individual predisposition to disease, and if we consider the great influence that environmental factors play in the development and progression of atherosclerosis, epigenetics is presented as a key element in trying to expand our knowledge on individual predisposition to atherosclerosis and cardiovascular disease.
Epigenetics can be described as the discipline that studies the mechanisms of transcriptional regulation, independent of changes in the sequence of DNA, and mostly induced by environmental factors.
This review aims to describe what epigenetics is and how epigenetic mechanisms are involved in atherosclerosis.
Los estudios de asociación basados en genes candidato llevados a cabo durante décadas han servido para visualizar la influencia del componente genético en enfermedades complejas como la arteriosclerosis. Arrojan como balance la identificación de múltiples variaciones con un efecto individual generalmente muy modesto. Aun presentando la notable limitación de depender de nuestro incompleto conocimiento de los procesos fisiológicos y fisiopatológicos, han servido también para evidenciar la interacción entre diferentes genes y, especialmente, la de estos con factores ambientales. En la última década, el desarrollo de los Genome-Wide Association Studies (GWAS) se contemplaba como una posibilidad de avance muy significativa ya que permitiría llevar a cabo estudios de asociación a escala genómica sin depender de la elección de genes candidato. Si bien los GWAS han identificado un buen número de nuevos genes candidato, ha supuesto una relativa sorpresa el hecho de que la mayoría de los SNP no se encuentren en zonas codificantes y que la mayoría de los nuevos genes identificados no guarden una relación funcional evidente con la enfermedad cardiovascular1. Ello nos indica que, tanto desde un punto de vista genómico como fisiopatológico, nuestro conocimiento de la enfermedad cardiovascular es aún limitado. Por lo tanto, queda aún camino por recorrer para descifrar la predisposición individual a la enfermedad, la llamada «missing heritability» (causas de la transmisión de predisposición a la enfermedad cardiovascular, aún por identificar)2. Si además tenemos en cuenta la gran influencia que los factores ambientales juegan en el desarrollo y la progresión de la arteriosclerosis, la epigenética se nos presenta como un elemento clave para intentar ampliar nuestro conocimiento de la predisposición individual a la arteriosclerosis y la enfermedad cardiovascular. Esta revisión pretende describir qué es la epigenética y de qué manera los mecanismos epigenéticos participan en la arteriosclerosis.
DefiniciónEl ADN contiene codificada la información necesaria para la transcripción de nuestros genes. Los genes no están siempre expresados y no se expresan por igual en todos los tejidos. Por lo tanto, el ADN no siempre se transcribe a ARN, y el ARN no siempre se traduce a proteínas. La complejidad de la regulación génica puede explicarse en parte gracias a la epigenética.
Existen varias definiciones de la epigenética pero la que probablemente mejor refleja su papel es que es la disciplina que estudia los procesos o modificaciones reversibles y heredables implicados en la regulación de la transcripción génica y que no dependen de cambios en la secuencia del ADN.
Estructura de la cromatinaEn el núcleo celular el ADN está empaquetado muy eficientemente y el grado de empaquetamiento determina la forma en que este ADN funcionará.
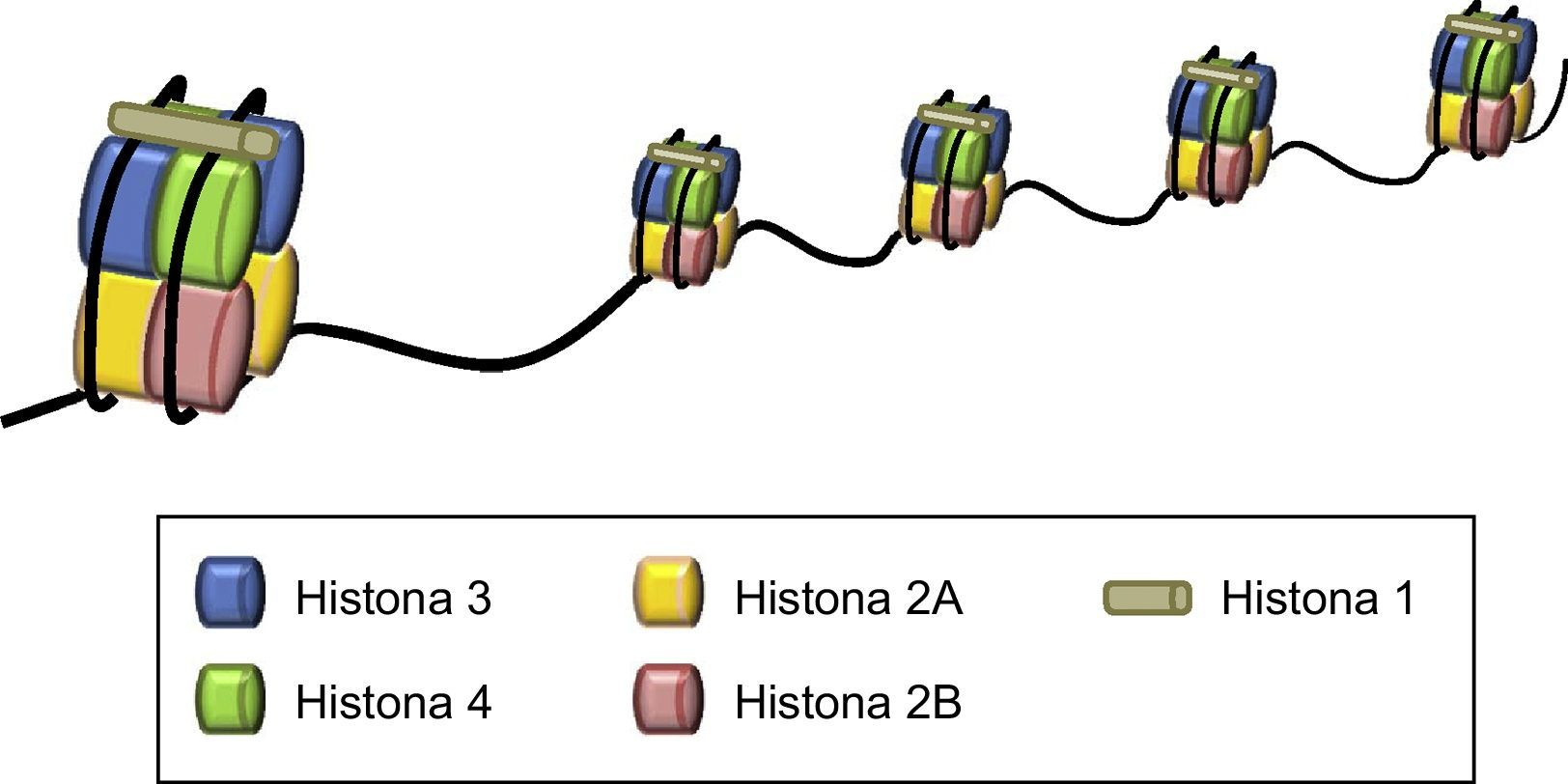
La cromatina es el primer nivel de empaquetamiento del ADN y representa el complejo formado por el ADN y las proteínas histonas organizadas formando un polímero cuya unidad básica son los nucleosomas, de manera que 146 pares de bases de ADN envuelven un octámero de histonas, formado por un tetrámero de las histonas H3 y H4 y 2 dímeros de H2A y H2B. La histona H1 mantiene lo nucleosomas unidos sellando los giros completos del ADN (fig. 1).

A su vez, la cromatina está organizada estructuralmente formando la heterocromatina o la eucromatina. La heterocromatina es una cromatina muy compacta y corresponde a la mayor parte del material nuclear. La heterocromatina incluye los telómeros y regiones pericentroméricas que son regiones que tienden a ser ricas en secuencias repetitivas y que tienen un bajo contenido génico. En cambio, el resto del genoma está formado por la eucromatina que es una estructura más laxa y transcripcionalmente activa y que contiene la mayoría de los genes.
Modificaciones epigenéticasLas modificaciones epigenéticas que sufren el ADN y la cromatina suelen asociarse a diferentes estados de la cromatina que en último término deciden qué funciones están activas y cuáles inactivas en cada tejido específico.
Metilación (e hidroximetilación) del ADNLa metilación del ADN es una de las modificaciones epigenéticas más estudiadas.
La metilación del ADN es un proceso activo y dinámico que tiene lugar gracias a una familia de enzimas, las ADN metiltranferasas (DNMT), que transfieren el grupo metilo desde la S-adenosil-metionina (SAM) al carbono 5′ de las citosinas.
La metilación del ADN ocurre principalmente en las citosinas localizadas en los dinucleótidos CpG. Los dinucleótidos CpG suelen estar repartidos heterogéneamente por todo el genoma, pero las zonas del genoma en las que la frecuencia de estos dinucleótidos CpG es unas 10 veces mayor en comparación con la media del genoma se conocen como islas CpG3. Típicamente una isla CpG tiene al menos 200 pares de bases con más del 50% de contenido de guaninas y citosinas y una proporción de CpG observada respecto a la esperada mayor del 60%. Las islas CpG suelen localizarse cerca de los sitios de inicio de la transcripción y por eso suelen asociarse con la región promotora de los genes. Ello llevó a pensar que podían tener un papel clave en la regulación de la transcripción génica. La metilación de las islas CpG se ha asociado a represión de los genes debido a que la presencia de citosinas metiladas produce un cambio conformacional en la doble cadena del ADN que dificulta la unión de factores de transcripción e incluso la propia transcripción.
Los promotores de los genes están normalmente desmetilados y juegan un papel clave al controlar la regulación génica4.
El perfil de metilación del ADN y la regulación de la expresión génica son específicos del tejido y de la célula.
Recientes evidencias indican que la metilación de las citosinas también puede tener lugar en las regiones adyacentes a las islas CpG, la denominada «orilla» (a 2kb flanqueando las islas CpG) e incluso que no está limitada a los dinucleótidos CpG, sino que también se ha descrito en los dinucleótidos CpA.
El patrón de metilación en un individuo se establece durante el desarrollo embrionario.
Mientras se forma el cigoto tiene lugar una desmetilación del ADN de las células germinales, y la metilación se reemprende durante las divisiones celulares necesarias del desarrollo (también se conoce como reprogramación epigenética). En este proceso participan las DNMT3A y DNMT3B que son metiltransferasas de novo, responsables de establecer el patrón de metilación de citosinas en el ADN no metilado5.
Una vez establecida la reprogramación, los patrones de metilación del ADN deberán ser mantenidos de forma estable por medio de las divisiones celulares. Esta función de mantenimiento se lleva a cabo gracias a la DNMT1, debido a su preferencia por el ADN hemimetilado y a su asociación estable con el ADN recién replicado, de tal manera que copia los patrones de metilación de las cadenas parentales en las cadenas recién sintetizadas durante la replicación del ADN.
El grado de metilación de un ADN también viene establecido por la desmetilación. Este proceso no está tan bien estudiado. La desmetilación puede tener lugar de forma pasiva durante la replicación del ADN o de forma activa, eliminando grupos metilo durante la fertilización.
La hidroximetilación es un paso intermedio de la desmetilación activa del ADN. Es una oxidación de la metilación mediada gracias a la familia de enzimas ten-eleven-translocation y también ocurre en la posición 5′ de las citosinas. Los niveles de hidroximetilación se han correlacionado con genes activos transcripcionalmente, al contrario de lo que mayoritariamente ocurre con la metilación6.
La hidroximetilación se ha descrito en todos los tejidos y tipos celulares estudiados hasta la fecha, aunque en mayor cantidad se ha descrito en el cerebro7. Mientras el porcentaje de citosinas metiladas a lo largo de diferentes tejidos está entre el 4 y 5% con relación a las guaninas, en el cerebro el 0,4-0,9% de las citosinas presentan hidroximetilación, y este porcentaje baja hasta el 0,2% en el riñón y 0,03% en el timo o los testículos8.
Respecto a la hidroximetilación se ha indicado que, además de marcador intermediario de la desmetilación, también puede servir como una nueva marca epigenética.
Modificaciones de histonasEl grado de empaquetamiento de los nucleosomas en la cromatina puede afectar la actividad transcripcional del ADN. Varias modificaciones epigenéticas que afectan tanto al ADN (metilación) como a las proteínas histonas (modificaciones postraduccionales debidas a metilación, acetilación, fosforilación o ubiquitinización) participan en el estado de compactación de la cromatina.
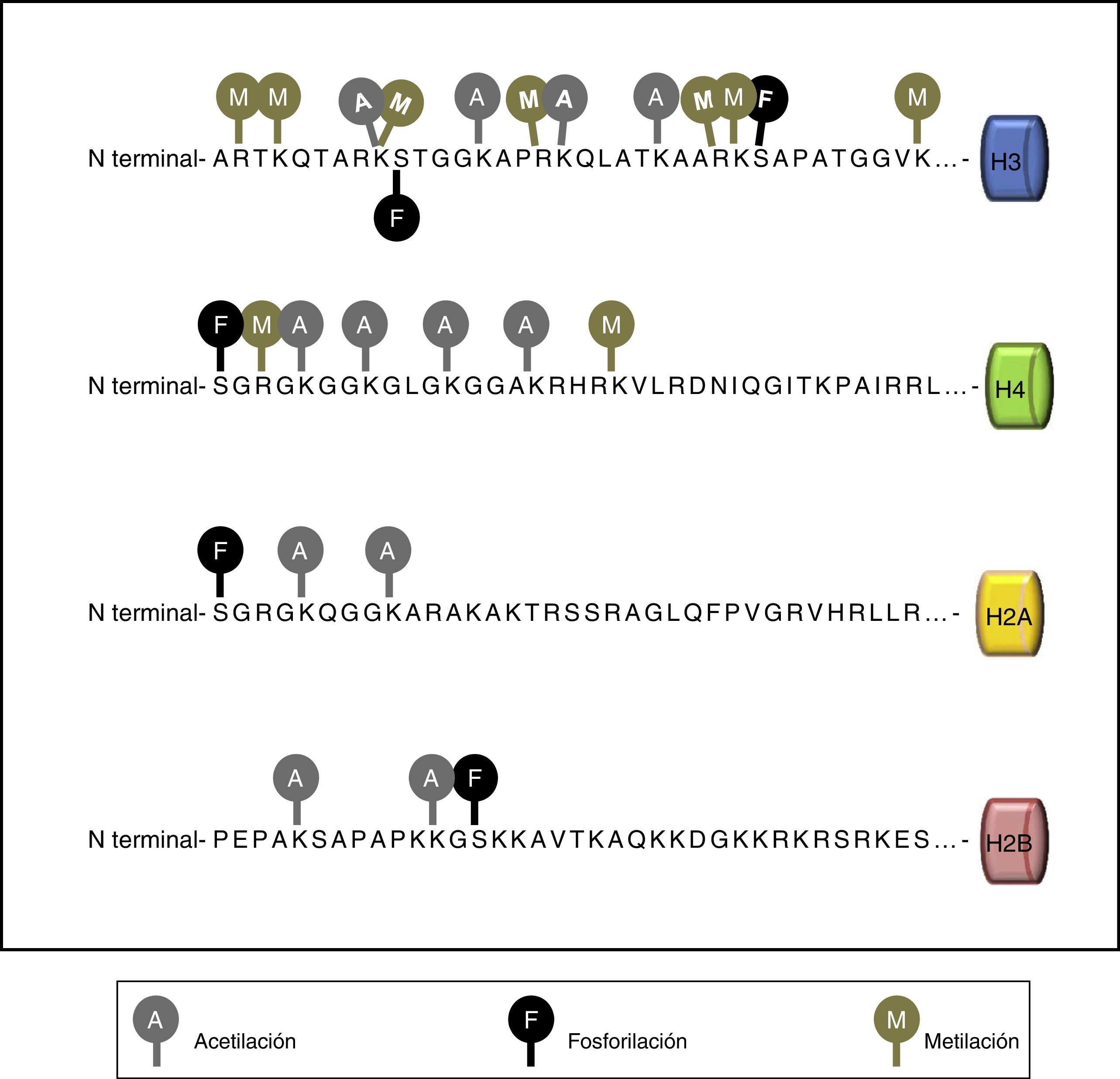
Las histonas tienen un dominio carboxilo terminal globular y una cola amino terminal que es la que sufre las modificaciones. Las modificaciones de las histonas incluyen metilación en los residuos de lisina (abreviada como K) y arginina (R), acetilación en residuos de lisina, ubiquitinación y sumoilación de lisinas y fosforilación de serinas (S) y treoninas (T).
Las modificaciones postraduccionales de las histonas tienen efectos directos sobre la estructura de la cromatina y, por lo tanto, sobre la regulación de la expresión génica.
Por ejemplo, la acetilación de lisinas se relaciona normalmente con un estado activo transcripcionalmente, mientras que la metilación de la arginina o la lisina resulta en activación o represión dependiendo de la posición del residuo en la cadena de la histona. Estas modificaciones pueden generar interacciones sinérgicas o antagonistas con proteínas asociadas a la cromatina, modulando el acceso al ADN o reclutando activamente reguladores transcripcionales.
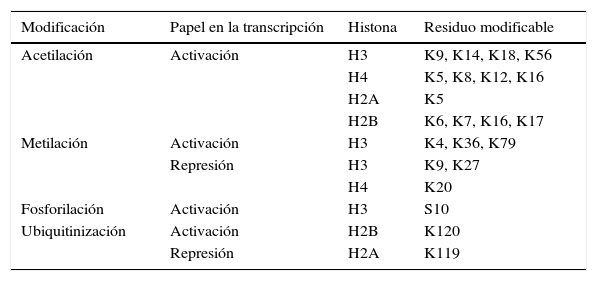
Las principales marcas de modificaciones de histonas están resumidas en la tabla 1 y la fig. 2.
Principales modificaciones en las histonas
| Modificación | Papel en la transcripción | Histona | Residuo modificable |
|---|---|---|---|
| Acetilación | Activación | H3 | K9, K14, K18, K56 |
| H4 | K5, K8, K12, K16 | ||
| H2A | K5 | ||
| H2B | K6, K7, K16, K17 | ||
| Metilación | Activación | H3 | K4, K36, K79 |
| Represión | H3 | K9, K27 | |
| H4 | K20 | ||
| Fosforilación | Activación | H3 | S10 |
| Ubiquitinización | Activación | H2B | K120 |
| Represión | H2A | K119 |
El número se refiere al número de aminoácido en la cadena de la histona.
K: residuo de lisina, S: residuo de serina.

El grado de acetilación de las histonas es un proceso modulado por las enzimas acetilasa de histonas (HAT) y deacetilasa de histonas (HDAC) y, en el caso del grado de metilación de las histonas, el proceso está mediado por las enzimas metiltransferasa de histonas (HMT) y demetilasa de histonas (HDM).
Como ya hemos comentado, la metilación del ADN y las modificaciones de las histonas son las principales marcas epigenéticas y las más estudiadas sobre todo con relación al desarrollo de cáncer, pero también son responsables de regular procesos implicados en la patogenia de la arteriosclerosis.
En células endoteliales se ha demostrado que las LDL oxidadas inducen la acetilación de la histona 4, y la fosforilación y acetilación de la histona 3 (marcas epigenéticas asociadas a activación de la expresión génica), y muestran como estas modificaciones de histonas son responsables de aumentar la expresión de citocinas proinflamatorias como IL8 y MCP1. Este proceso se puede controlar gracias al tratamiento previo de las células endoteliales con estatinas9.
El butirato es un ácido graso de cadena corta subproducto de la digestión de la fibra dietética y también es un conocido inhibidor dietético de HDAC que inhibe la proliferación de las células musculares lisas vasculares (CMLV) y, recientemente, se ha descrito que este proceso tiene lugar a través de modificaciones en las histonas, más concretamente acetilando, fosforilando y metilando residuos aminoácido de la histona 3. Estas modificaciones modulan la expresión de genes implicados en el ciclo celular y reprimen la proliferación de las CMLV10.
En un modelo animal de arteriosclerosis, con un ratón deficiente en apoE, se describió que las células musculares lisas están hipometiladas globalmente en la lesión arteriosclerótica11, y que la arteriosclerosis se asocia con un reajuste de los patrones de metilación ya sea por hipo- o por hipermetilación, en los diferentes tejidos diana en el desarrollo de la arteriosclerosis12.
Comparando el grado de metilación en dinucleótidos CpG repartidos a lo largo de todo el genoma de aortas sanas y aortas afectas de arteriosclerosis obtenidas post mórtem del mismo individuo, Zaina et al.13 han descrito que en las aortas con lesión arteriosclerótica hay un patrón de metilación del ADN global alterado de manera que encuentran más regiones hipermetiladas en las aortas afectas. Analizando en detalle las zonas diferencialmente metiladas en arteriosclerosis, han identificado CpG localizados en varios genes implicados en la homeostasis vascular controlando, por ejemplo, la migración de monocitos (PDGFD y MYH10); estimulando la angiogénesis (MIR23b y HOXB3); controlando la proliferación de las CMLV (RPTOR), su señalización (PTK2, EGFR, TSC22D1 y PRKCE) y su contracción (KCNMA1 y CALD1); y participando en el estado y calcificación de la placa (FBN2, A2BP1 y PLA2G10).
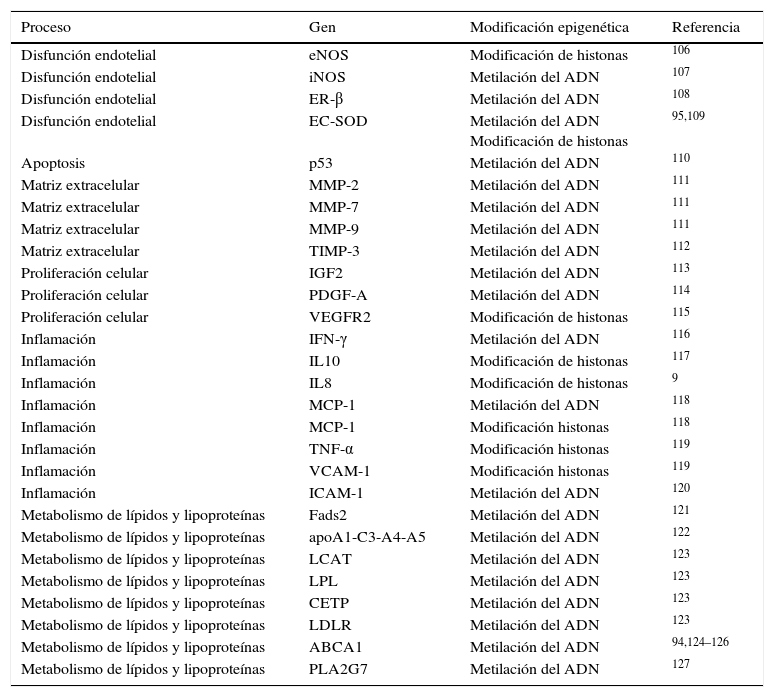
Existen otros ejemplos en la bibliografía que han descrito cambios en el patrón de metilación del ADN o de modificaciones en histonas, en genes implicados en procesos clave en el desarrollo de arteriosclerosis como son la disfunción endotelial, el control de la inflamación y oxidación, y el metabolismo de los lípidos, entre otros (resumidos en la tabla 2).
Genes relacionados con la arteriosclerosis que son regulados epigenéticamente
| Proceso | Gen | Modificación epigenética | Referencia |
|---|---|---|---|
| Disfunción endotelial | eNOS | Modificación de histonas | 106 |
| Disfunción endotelial | iNOS | Metilación del ADN | 107 |
| Disfunción endotelial | ER-β | Metilación del ADN | 108 |
| Disfunción endotelial | EC-SOD | Metilación del ADN Modificación de histonas | 95,109 |
| Apoptosis | p53 | Metilación del ADN | 110 |
| Matriz extracelular | MMP-2 | Metilación del ADN | 111 |
| Matriz extracelular | MMP-7 | Metilación del ADN | 111 |
| Matriz extracelular | MMP-9 | Metilación del ADN | 111 |
| Matriz extracelular | TIMP-3 | Metilación del ADN | 112 |
| Proliferación celular | IGF2 | Metilación del ADN | 113 |
| Proliferación celular | PDGF-A | Metilación del ADN | 114 |
| Proliferación celular | VEGFR2 | Modificación de histonas | 115 |
| Inflamación | IFN-γ | Metilación del ADN | 116 |
| Inflamación | IL10 | Modificación de histonas | 117 |
| Inflamación | IL8 | Modificación de histonas | 9 |
| Inflamación | MCP-1 | Metilación del ADN | 118 |
| Inflamación | MCP-1 | Modificación histonas | 118 |
| Inflamación | TNF-α | Modificación histonas | 119 |
| Inflamación | VCAM-1 | Modificación histonas | 119 |
| Inflamación | ICAM-1 | Metilación del ADN | 120 |
| Metabolismo de lípidos y lipoproteínas | Fads2 | Metilación del ADN | 121 |
| Metabolismo de lípidos y lipoproteínas | apoA1-C3-A4-A5 | Metilación del ADN | 122 |
| Metabolismo de lípidos y lipoproteínas | LCAT | Metilación del ADN | 123 |
| Metabolismo de lípidos y lipoproteínas | LPL | Metilación del ADN | 123 |
| Metabolismo de lípidos y lipoproteínas | CETP | Metilación del ADN | 123 |
| Metabolismo de lípidos y lipoproteínas | LDLR | Metilación del ADN | 123 |
| Metabolismo de lípidos y lipoproteínas | ABCA1 | Metilación del ADN | 94,124–126 |
| Metabolismo de lípidos y lipoproteínas | PLA2G7 | Metilación del ADN | 127 |
La finalización del Proyecto Genoma Humano en 2003 permitió identificar que solo menos del 2% del genoma humano se utiliza para codificar proteínas. La mayoría del genoma se transcribe activamente para producir tránscritos no codificantes. Los ARN no-codificantes son moléculas de ARN funcionales que, aunque no codifican para proteínas, son clave en la regulación génica.
La transcripción del genoma en diferentes formas de ARN, incluyendo los ARN no codificantes (ARNnc), está bien establecida en la actualidad. Los ARNnc se consideran marcas epigenéticas debido a que participan en el control de la expresión génica sin implicar cambios en la secuencia de nucleótidos o en el número de copias del ADN. Sin embargo, el mecanismo de acción de los ARNnc difiere del que se ha descrito anteriormente para las otras marcas epigenéticas (metilación del ADN, modificación de histonas). Así, los ARNnc no producen cambios sobre la molécula de ADN sino que se encargan de controlar la expresión génica de manera postranscripcional uniéndose a ARN mensajeros (ARNm) diana y, de esta forma, promoviendo o bien su degradación, o inhibiendo su traducción, evitando en los 2 casos la expresión génica. De esta forma los ARNnc reguladores pueden contribuir de manera importante en el control epigenético de la expresión génica.
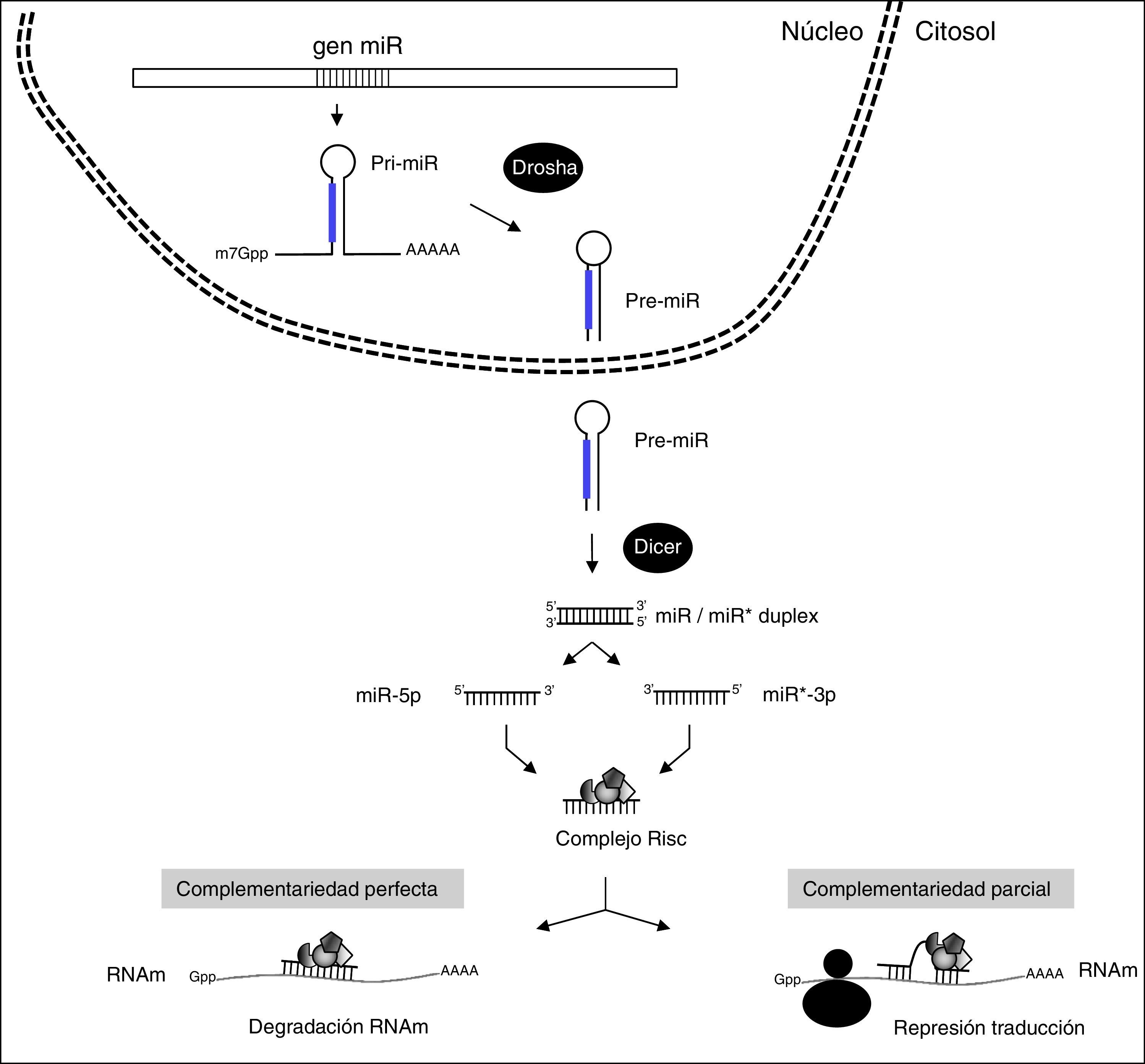
Los ARNnc mejor estudiados y caracterizados son los microARN (miR) que se encargan de ajustar y dirigir finamente la traducción de hasta un 60% de los genes codificantes de proteínas en humanos14. Los miR maduros y funcionales son moléculas pequeñas de ARN de cadena sencilla de aproximadamente 22 nucleótidos que están codificados en el genoma y cuya biogénesis está altamente regulada (fig. 3). Así, los genes codificantes de los miR se transcriben inicialmente como miR primarios (pri-miR) de cadena larga por acción de la ARN polimerasa III. Estos son procesados en el núcleo por un complejo de proteínas, entre las que destaca Drosha, para formar estructuras de aproximadamente 70 nucleótidos denominadas miR precursores (pre-miR). Los pre-miR resultantes son seguidamente transportados al citoplasma a través de exportina 5 donde son procesados por una segunda RNasa III denominada Dicer que genera complejos de doble cadena del tamaño de los miRs maduros (22 nucleótidos). De las 2 cadenas del complejo preferentemente se selecciona una como miR maduro, conocida como cadena guía, para integrarse en el complejo RNA-induced silencing complex (RISC) encargado de producir el silenciamiento génico, mientras que la otra cadena, denominada pasajera, normalmente se degrada. Sin embargo, en muchos casos las 2 cadenas son biológicamente activas y funcionan como miR diferentes. En este caso los 2 miR maduros provienen del mismo pre-miR y son denominados como 5p y 3p en función de la cadena a partir de la cual se generan. El complejo RISC-miR se une específicamente a regiones complementarias situadas en la zona 3′ UTR de los ARNm diana, promoviendo principalmente su desestabilización y degradación cuando la unión es por complementariedad perfecta o, en ocasiones, su represión de la traducción cuando la unión es por complementariedad parcial (fig. 3). La especificidad y funcionalidad de los miR están determinadas por la llamada región «semilla» del miR constituida por los nucleótidos 2 a 7 de la región 5′ de cada miR maduro. Esta región debe ser obligatoriamente complementaria al ARNm diana15 mientras que el resto puede tener complementariedad parcial. Este hecho permite que los miR individuales puedan actuar inhibiendo múltiples ARNm16 de manera que la alteración de un solo miR puede resultar en diferentes fenotipos. Además, un ARNm individual puede contener diferentes dianas de unión para distintos miR, resultando todo ello en una compleja red reguladora17.

Además de los miR, existen los ARN no codificantes largos (lncARN del inglés) formados por más de 200 nucleótidos que también funcionan como reguladores epigenéticos importantes y que en diferentes estudios se han descrito como reguladores del fenotipo vascular18,19. Esta regulación se lleva a cabo básicamente dentro del núcleo celular mientras que el control por parte de los miR es básicamente citoplasmático20. Entre los lncARN podemos destacar Fendrr, CHRF, Anril, o Malat1 que regulan procesos tales como la expresión génica cardíaca, la diferenciación de miocitos, la hipertrofia cardíaca, la angiogénesis o la susceptibilidad a enfermedad cardiovascular21–24.
Recientemente se ha añadido un nivel más de complejidad al demostrarse la regulación epigenética de las marcas epigenéticas, es decir, se ha descrito una intercomunicación entre mecanismos epigenéticos. Concretamente, metilaciones en islas CpG de regiones reguladoras de miR así como modificaciones en histonas se han implicado en la patogénesis de diferentes enfermedades25–29 alterando la expresión de miR y regulando los mARN diana30–33. Además los mismos miR son capaces de controlar la metilación del ADN y la modificación de histonas mediante la regulación de la expresión de genes que controlan estas rutas epigenéticas. Así, se ha descrito que miR-1 promueve la miogénesis actuando sobre HDAC4 y miR-29 tiene como diana la ADN metiltransferasa DNMT3A25,29,34. Es interesante destacar que estos cambios epigenéticos pueden ser causados por una variedad de factores tales como químicos, fármacos, edad y dieta35.
La intercomunicación entre miR y los factores de transcripción clásicos también está bien establecida ya desde los primeros estudios de predicción de las dianas de los miR cuando se estableció la preferencia de estos factores de transcripción como dianas de actuación de los miR36. Además, la expresión de los miR puede estar regulada por factores de transcripción, de manera que se crean circuitos de control con regulación positiva o negativa37.
En la actualidad hay descritos más de 1.000 miR en humanos. En los últimos años se ha ido describiendo el papel importante que estos juegan en las enfermedades cardiovasculares, no solo como moléculas implicadas en la patogenia de los diferentes fenotipos cardiovasculares, sino que también como posibles biomarcadores de riesgo y progresión de enfermedades cardíacas como infarto agudo de miocardio, enfermedad cardiovascular o insuficiencia cardíaca18,38–53. En este sentido, un resultado importante que explica la función crucial de los miR en el desarrollo del sistema cardiovascular es el que describe que la eliminación en ratones de Dicer (la RNasa que procesa los pre-miR) conlleva defectos en angiogénesis, formación de vasos y desarrollo cardíaco54.
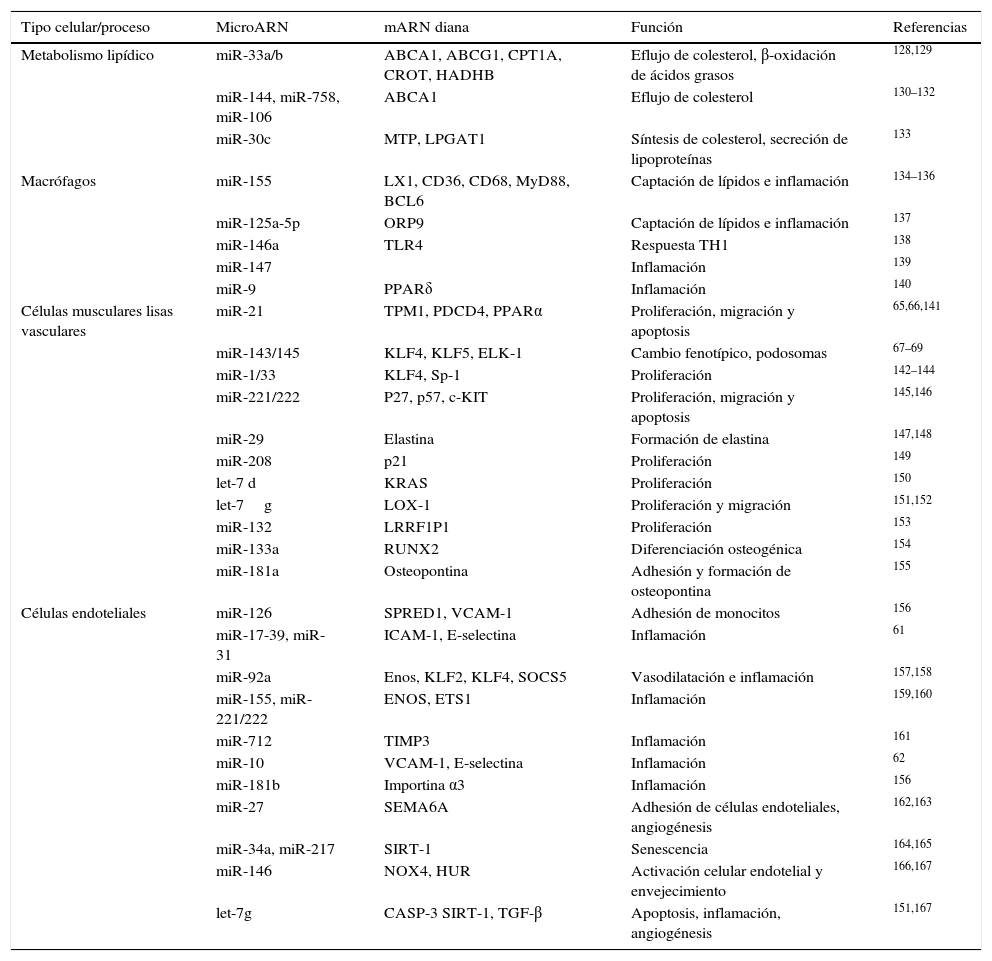
Micro ARN y arteriosclerosisEl interés en el estudio de los miR ha permitido que se hayan relacionado con muchos de los procesos o mecanismos básicos implicados en el desarrollo de arteriosclerosis. Así, se ha demostrado que los miR funcionan como controladores clave de la expresión génica, y, por lo tanto, de la funcionalidad, de los diferentes tipos celulares implicados en la arteriosclerosis (células endoteliales, células musculares lisas vasculares, monocitos y macrófagos). Además, se han implicado en el control de procesos tales como la inflamación, el metabolismo cardíaco y lipoproteico, la biosíntesis y captación de colesterol, el remodelaje cardíaco, la disfunción endotelial, la angiogénesis o en la diferenciación, migración y proliferación celular. Una exhaustiva relación de todos los miR implicados en estos procesos así como de los detalles de los genes diana y sus mecanismos han sido magníficamente revisados muy recientemente y están resumidos en la tabla 355. Así, se ha descrito que tanto miR-33a como miR-324 reprimen la expresión de diferentes genes implicados en el eflujo de colesterol como por ejemplo ABCA1, de manera que tanto la inhibición de miR-33, como el déficit del gen en ratones aumenta los niveles de colesterol HDL, atenúan la progresión de arteriosclerosis y aumentan su regresión56–58. Además, en ratones deficientes de apoE, la ausencia de miR-33 reduce marcadamente la lesión arteriosclerótica que desarrollan estos ratones. También se ha descrito que la sobreexpresión hepática de miR-30c en ratones produce una reducción importante del colesterol plasmático, mejorando así la progresión de arteriosclerosis59. Por otra parte, también se han descrito efectos de miR sobre el endotelio vascular. Así se ha demostrado que la administración de miR-126 en ratones reduce el tamaño de la placa arteriosclerótica a través de disminuir el contenido en macrófagos y células apoptóticas debido a una disminución de VCAM endotelial60. De forma similar, miR-31, miR-17-3p y miR-21 regulan en células endoteliales los niveles de VCAM-1, ICAM-1 y E-selectina61. Otras señales proinflamatorias como la ruta de señalización de NF-κB están inhibidas por miR-10a62 y miR-181b63, que actúan sobre las subunidades encargadas de translocar NF-κB al núcleo. En concreto, la inhibición de importina α3 por parte de miR-181b resulta en una reducción de la arteriosclerosis presente en ratones deficientes de apoE64. Por otra parte, los miR regulan tanto la diferenciación como la proliferación de las células musculares lisas, procesos clave en el desarrollo de arteriosclerosis. Así, miR-21 y miR-143/145 regulan la diferenciación de estas células actuando respectivamente sobre tropomiosina 165 o PPARα66 y sobre KLF4, ELK-1 o Krüppel-like factor67–70.
MicroARN implicados en el desarrollo de la arteriosclerosis
| Tipo celular/proceso | MicroARN | mARN diana | Función | Referencias |
|---|---|---|---|---|
| Metabolismo lipídico | miR-33a/b | ABCA1, ABCG1, CPT1A, CROT, HADHB | Eflujo de colesterol, β-oxidación de ácidos grasos | 128,129 |
| miR-144, miR-758, miR-106 | ABCA1 | Eflujo de colesterol | 130–132 | |
| miR-30c | MTP, LPGAT1 | Síntesis de colesterol, secreción de lipoproteínas | 133 | |
| Macrófagos | miR-155 | LX1, CD36, CD68, MyD88, BCL6 | Captación de lípidos e inflamación | 134–136 |
| miR-125a-5p | ORP9 | Captación de lípidos e inflamación | 137 | |
| miR-146a | TLR4 | Respuesta TH1 | 138 | |
| miR-147 | Inflamación | 139 | ||
| miR-9 | PPARδ | Inflamación | 140 | |
| Células musculares lisas vasculares | miR-21 | TPM1, PDCD4, PPARα | Proliferación, migración y apoptosis | 65,66,141 |
| miR-143/145 | KLF4, KLF5, ELK-1 | Cambio fenotípico, podosomas | 67–69 | |
| miR-1/33 | KLF4, Sp-1 | Proliferación | 142–144 | |
| miR-221/222 | P27, p57, c-KIT | Proliferación, migración y apoptosis | 145,146 | |
| miR-29 | Elastina | Formación de elastina | 147,148 | |
| miR-208 | p21 | Proliferación | 149 | |
| let-7 d | KRAS | Proliferación | 150 | |
| let-7g | LOX-1 | Proliferación y migración | 151,152 | |
| miR-132 | LRRF1P1 | Proliferación | 153 | |
| miR-133a | RUNX2 | Diferenciación osteogénica | 154 | |
| miR-181a | Osteopontina | Adhesión y formación de osteopontina | 155 | |
| Células endoteliales | miR-126 | SPRED1, VCAM-1 | Adhesión de monocitos | 156 |
| miR-17-39, miR-31 | ICAM-1, E-selectina | Inflamación | 61 | |
| miR-92a | Enos, KLF2, KLF4, SOCS5 | Vasodilatación e inflamación | 157,158 | |
| miR-155, miR-221/222 | ENOS, ETS1 | Inflamación | 159,160 | |
| miR-712 | TIMP3 | Inflamación | 161 | |
| miR-10 | VCAM-1, E-selectina | Inflamación | 62 | |
| miR-181b | Importina α3 | Inflamación | 156 | |
| miR-27 | SEMA6A | Adhesión de células endoteliales, angiogénesis | 162,163 | |
| miR-34a, miR-217 | SIRT-1 | Senescencia | 164,165 | |
| miR-146 | NOX4, HUR | Activación celular endotelial y envejecimiento | 166,167 | |
| let-7g | CASP-3 SIRT-1, TGF-β | Apoptosis, inflamación, angiogénesis | 151,167 |
Además, se ha descrito que una expresión génica anómala asociada a niveles alterados de miR es un determinante de muchos fenotipos patológicos tales como dislipidemia, resistencia a insulina, obesidad o diabetes17. En estos casos los miR actúan regulando el metabolismo en los órganos responsables de estos procesos tales como el páncreas, el hígado o el tejido adiposo17.
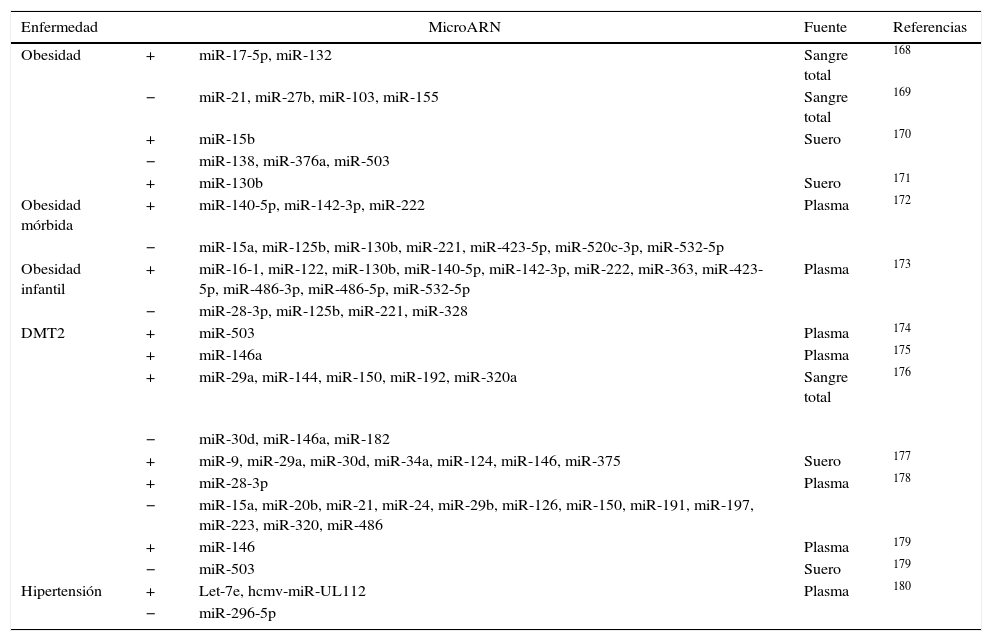
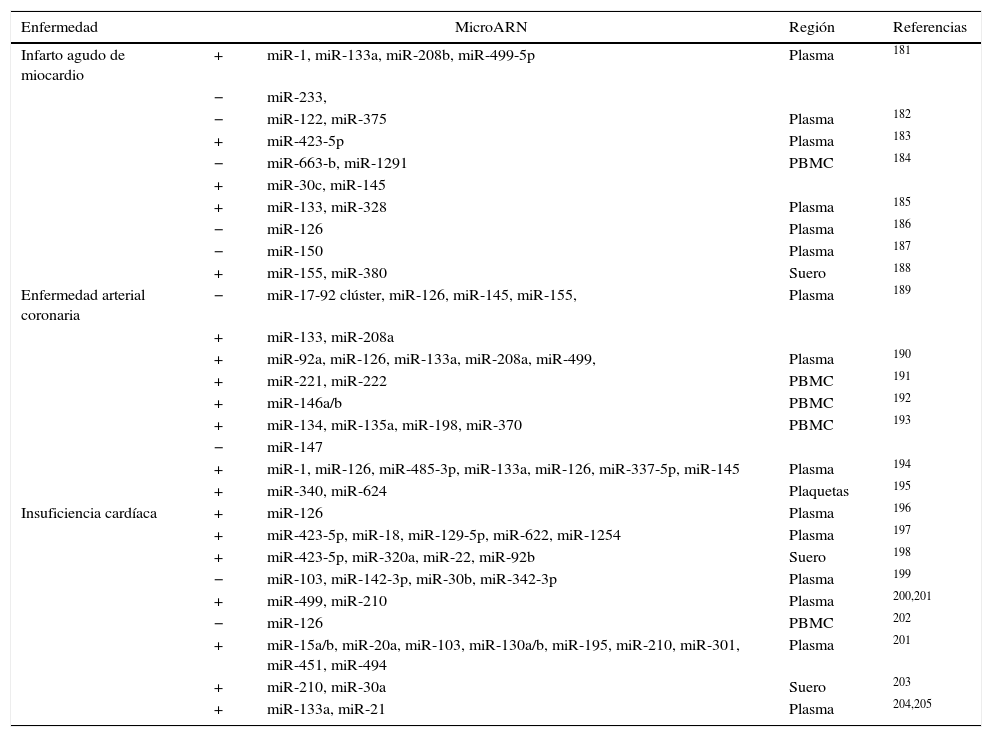
Micro ARN circulantesAdemás de regular dianas intracelulares, se ha descrito que los miR están presentes en sangre a concentraciones fisiológicamente significativas. A pesar de que los estímulos que activan la secreción de los miR a la circulación no están claros, los miR circulantes pueden transportarse de forma estable y ser entregados a células destino60,71 gracias a su incorporación a cuerpos apoptóticos60, exosomas72, a partículas de HDL73 y a la formación de complejos con argonauta 274. Se necesitan más estudios para clarificar si los miR endógenos circulantes pueden ser dirigidos a células diana para facilitar la comunicación entre diferentes órganos y regular así la expresión génica a nivel sistémico. Sí está mucho más estudiado el papel de los miR circulantes como biomarcadores específicos de enfermedad. Los niveles de expresión en sangre o en plasma de miR específicos están alterados de manera significativa en diferentes enfermedades cardiovasculares como el infarto agudo de miocardio o la insuficiencia cardíaca, así como en obesidad, hipertensión y diabetes mellitus tipo 2 (DMT2). El reto en este tipo de estudios está en identificar de forma precisa y reproducible miR específicos o un conjunto de ellos que puedan utilizarse como biomarcadores de riesgo o de progresión de enfermedad y que permitan identificar a aquellos pacientes susceptibles de desarrollar la enfermedad. Existen muchos estudios que han planteado este objetivo aunque en la actualidad los resultados son poco consistentes entre ellos. En la tabla 4 adaptada de Price et al.17 se resumen los miR más significativos asociados con obesidad, DMT2 e hipertensión y en la tabla 5 se detallan aquellos implicados en infarto agudo de miocardio, en enfermedad coronaria arterial e insuficiencia cardíaca. El papel de los miR en esta última enfermedad se ha estudiado extensamente. En los últimos años diferentes estudios han demostrado una función importante de los miR en la patogénesis de la insuficiencia cardíaca. Por ejemplo se ha propuesto un patrón de expresión de miR específico para diferentes etiologías de insuficiencia cardíaca. Así, 43 miR estaban diferencialmente expresados en miocardiopatía mientras que 13 eran específicos de estenosis aórtica75. La sobreexpresión in vitro de diferentes miR individuales que se han descrito sobreexpresados en HF (miR-23a, -23b, -24, -195 o -214) inducía hipertrofia cardíaca. La sobreexpresión de miR-133 que se ha descrito reprimida en HF inhibía la hipertrofia cardíaca, mientras que su supresión la promovía tanto in vivo como in vitro76,77.
MicroARN asociados a obesidad, DMT2 e hipertensión
| Enfermedad | MicroARN | Fuente | Referencias | |
|---|---|---|---|---|
| Obesidad | + | miR-17-5p, miR-132 | Sangre total | 168 |
| − | miR-21, miR-27b, miR-103, miR-155 | Sangre total | 169 | |
| + | miR-15b | Suero | 170 | |
| − | miR-138, miR-376a, miR-503 | |||
| + | miR-130b | Suero | 171 | |
| Obesidad mórbida | + | miR-140-5p, miR-142-3p, miR-222 | Plasma | 172 |
| − | miR-15a, miR-125b, miR-130b, miR-221, miR-423-5p, miR-520c-3p, miR-532-5p | |||
| Obesidad infantil | + | miR-16-1, miR-122, miR-130b, miR-140-5p, miR-142-3p, miR-222, miR-363, miR-423-5p, miR-486-3p, miR-486-5p, miR-532-5p | Plasma | 173 |
| − | miR-28-3p, miR-125b, miR-221, miR-328 | |||
| DMT2 | + | miR-503 | Plasma | 174 |
| + | miR-146a | Plasma | 175 | |
| + | miR-29a, miR-144, miR-150, miR-192, miR-320a | Sangre total | 176 | |
| − | miR-30d, miR-146a, miR-182 | |||
| + | miR-9, miR-29a, miR-30d, miR-34a, miR-124, miR-146, miR-375 | Suero | 177 | |
| + | miR-28-3p | Plasma | 178 | |
| − | miR-15a, miR-20b, miR-21, miR-24, miR-29b, miR-126, miR-150, miR-191, miR-197, miR-223, miR-320, miR-486 | |||
| + | miR-146 | Plasma | 179 | |
| − | miR-503 | Suero | 179 | |
| Hipertensión | + | Let-7e, hcmv-miR-UL112 | Plasma | 180 |
| − | miR-296-5p | |||
MicroARN asociados a infarto agudo de miocardio, enfermedad coronaria arterial e insuficiencia cardíaca
| Enfermedad | MicroARN | Región | Referencias | |
|---|---|---|---|---|
| Infarto agudo de miocardio | + | miR-1, miR-133a, miR-208b, miR-499-5p | Plasma | 181 |
| − | miR-233, | |||
| − | miR-122, miR-375 | Plasma | 182 | |
| + | miR-423-5p | Plasma | 183 | |
| − | miR-663-b, miR-1291 | PBMC | 184 | |
| + | miR-30c, miR-145 | |||
| + | miR-133, miR-328 | Plasma | 185 | |
| − | miR-126 | Plasma | 186 | |
| − | miR-150 | Plasma | 187 | |
| + | miR-155, miR-380 | Suero | 188 | |
| Enfermedad arterial coronaria | − | miR-17-92 clúster, miR-126, miR-145, miR-155, | Plasma | 189 |
| + | miR-133, miR-208a | |||
| + | miR-92a, miR-126, miR-133a, miR-208a, miR-499, | Plasma | 190 | |
| + | miR-221, miR-222 | PBMC | 191 | |
| + | miR-146a/b | PBMC | 192 | |
| + | miR-134, miR-135a, miR-198, miR-370 | PBMC | 193 | |
| − | miR-147 | |||
| + | miR-1, miR-126, miR-485-3p, miR-133a, miR-126, miR-337-5p, miR-145 | Plasma | 194 | |
| + | miR-340, miR-624 | Plaquetas | 195 | |
| Insuficiencia cardíaca | + | miR-126 | Plasma | 196 |
| + | miR-423-5p, miR-18, miR-129-5p, miR-622, miR-1254 | Plasma | 197 | |
| + | miR-423-5p, miR-320a, miR-22, miR-92b | Suero | 198 | |
| − | miR-103, miR-142-3p, miR-30b, miR-342-3p | Plasma | 199 | |
| + | miR-499, miR-210 | Plasma | 200,201 | |
| − | miR-126 | PBMC | 202 | |
| + | miR-15a/b, miR-20a, miR-103, miR-130a/b, miR-195, miR-210, miR-301, miR-451, miR-494 | Plasma | 201 | |
| + | miR-210, miR-30a | Suero | 203 | |
| + | miR-133a, miR-21 | Plasma | 204,205 | |
PBMC: células mononucleadas de sangre periférica.
Finalmente, está descrito que algunos miR (miR1, 133a y 499) pueden aumentar la proliferación y la diferenciación de cardiomiocitos humanos provenientes de células madre embrionarias o de células progenitoras humanas78,79. Además, utilizando estos miR específicos se puede aumentar la reprogramación de fibroblastos adultos directamente en células con linaje cardiomiocítico80. También se ha descrito que una sobreexpresión del clúster miR-17-92 en ratones aumenta la proliferación de células progenitoras cardíacas y facilita la regeneración cardíaca después de un infarto agudo de miocardio53. Así, es posible pensar que estos miR puedan convertirse en dianas terapéuticas para la reparación del daño cardíaco y la regeneración del corazón aunque para ello todavía es necesario mejorar la tecnología de entrega de estos miR a sus células diana para evitar inhibiciones o activaciones inespecíficas de otros miR y hacer frente a posibles problemas de seguridad.
Metilación del ARNDesde hace ya unos años se están describiendo modificaciones en el ARN que pueden funcionar como reguladoras de la expresión génica a nivel postraduccional, pero aún se conoce poco.
Una de las modificaciones más estudiadas es la metilación del ARN que puede tener lugar en diferentes posiciones en el nucleótido de tARN, mARN y rARN.
La metilación del ARN tiene diferentes consecuencias funcionales como estabilización, aumento en la función y control de calidad.
Uno de los principales residuos modificados en el mARN es la metilación en posición 6 de la adenosina (m6A), proceso en el que SAM también es el donador del grupo metilo, y se ha demostrado que es una modificación dinámica que juega un papel importante en obesidad y DMT281 al igual que en ciertos tipos de cáncer y enfermedades mentales.
Impronta genética y «memoria celular» en épocas tempranas de la vidaLa impronta es un mecanismo epigenético reversible especialmente importante en mamíferos. La impronta afecta la expresión de algunos, no todos, los genes, y se caracteriza por la expresión de solo uno de los alelos (el alelo que proviene de la madre o el que proviene del padre). El alelo inactivo es el imprintado y el silenciamiento tiene lugar principalmente mediante metilación de la región del ADN que contiene el gen imprintado.
La impronta tiene lugar en los gametos. En estas células, los patrones originales son borrados y solo uno específico, el materno o el paterno, es reestablecido.
En el gen imprintado solo uno de los alelos funciona. Por lo tanto, ante una situación de pérdida de función del alelo activo, este no podrá ser compensado por el otro.
Los genes imprintados tienen papeles clave en el desarrollo prenatal, en el comportamiento e incluso en el desarrollo de enfermedades en humanos como cáncer, obesidad, o diabetes. Por ejemplo, la administración de una dieta rica en grasas en ratones afectaba al peso corporal de la tercera generación de su descendencia y este efecto solo era observable en las hembras, lo que indica que la influencia ambiental sobre el desarrollo del peso corporal puede ser responsable de loci imprintados82.
Conocidos síndromes pediátricos son debidos a procesos de impronta, como el Prader-Willi o Angelman que son debidos a errores genéticos y epigenéticos en el cromosoma 15.
Aproximadamente unos 100 genes humanos están imprintados y la mayoría se localizan en los cromosomas 6, 7, 11, 14, 15 y 20.
Dieta y folato/homocisteínaEl grado de expresión de un gen ya hemos visto que viene determinado, en parte, por mecanismos epigenéticos, y estos pueden alterarse como consecuencia de influencias ambientales. Es decir que las interacciones gen-ambiente son mediadas por modificaciones epigenéticas, y los cambios epigenéticos suelen aparecer en respuesta a cambios en el ambiente como cambios en el estado nutricional, y esto es especialmente relevante en enfermedades complejas como el cáncer, la diabetes o la enfermedad cardiovascular, debido a que su desarrollo tiene un origen genético influido por un fuerte componente ambiental y de estilo de vida.
Los grupos metilos son adquiridos a través de la dieta y son donados al ADN a través de la vía del folato y de la metionina.
os factores dietéticos son necesarios para generar SAM que es el donador principal de grupos metilo. Los nutrientes ricos en grupos metilos incluyen: vitaminas como el folato, la riboflavina, vitamina B12, vitamina B6 y colina; y aminoácidos como la metionina, la cisteína, la glicina y la serina.
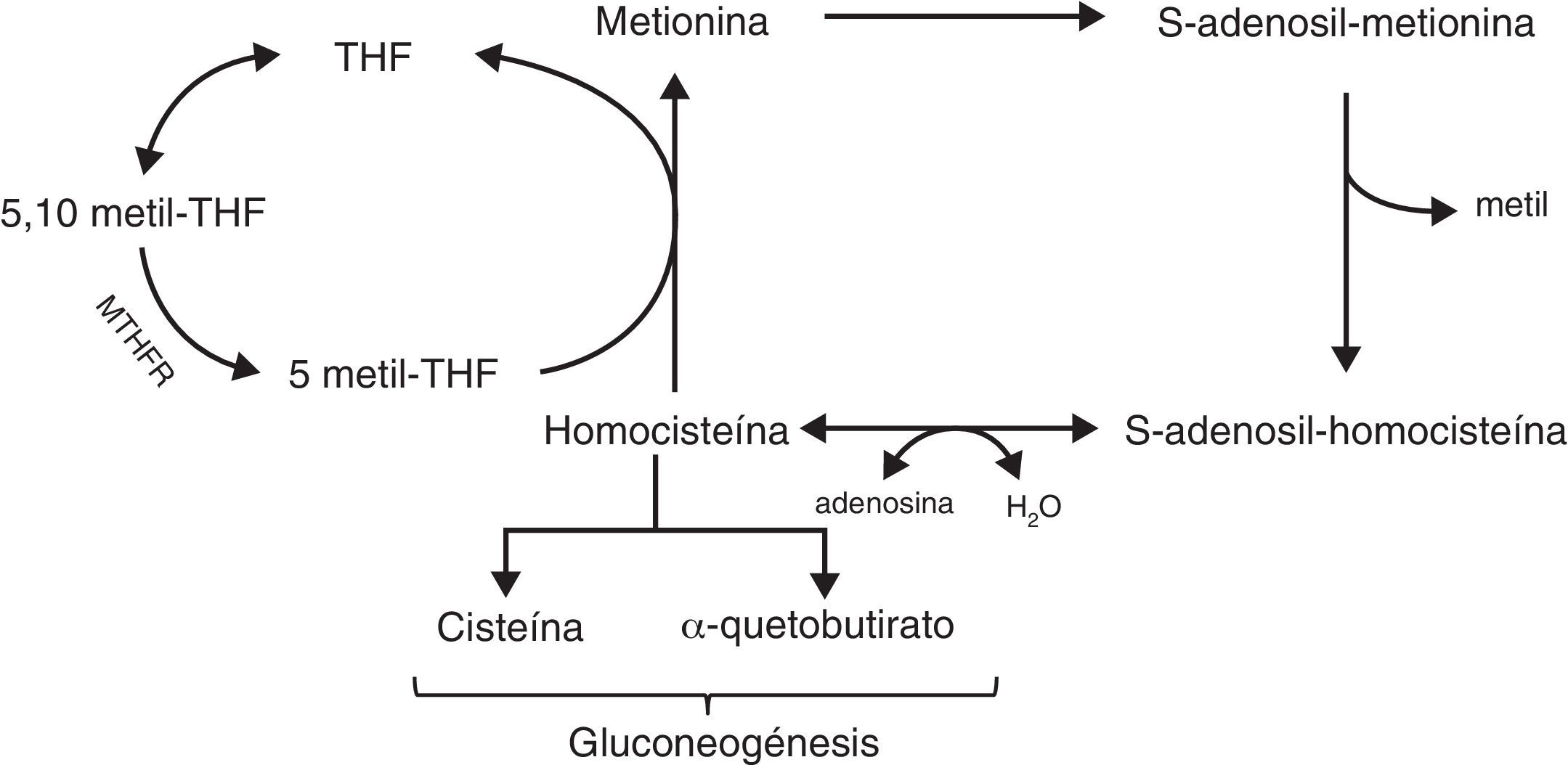
La formación de SAM implica 3 vías bioquímicas interrelacionadas: el ciclo del folato, el ciclo de la metionina y la vía de transulfuración (fig. 4)83.
Ciclo del folato
El folato es una vitamina B hidrosoluble cuya forma activa es el tetrahidrofolato (THF) que se utiliza en la formación de purinas y pirimidinas, y la remetilación de homocisteína a metionina.
Para la formación de purinas y pirimidinas el THF es convertido en 5,10-metil-THF que gracias a la MTHFR se convierte en 5-metil-THF.
Ciclo de la metioninaLa metionina es un aminoácido que se convierte en SAM (principal donador de grupos metilo al ADN, ARN, proteínas, fosfolípidos…) gracias a la metionin-adenosiltransferasa. Cuando SAM dona su metilo, se forma el S-adenosil-homocisteína (SAH) y, tras la liberación del grupo adenosil, obtenemos la homocisteína.
La homocisteína es un aminoácido no implicado en la síntesis de proteínas que puede ser remetilado a metionina gracias a la donación del grupo metilo por parte de la 5-metil-THF o puede ser catabolizado a cisteína a través de la vía de transulfuración.
Vía de la transulfuraciónLa homocisteína puede ser convertida en cisteína y α-quetobutirato que tras varias reacciones serán convertidos en piruvato y succinil CoA necesarios en la gluconeogénesis.
Con relación a la enfermedad cardiovascular, tanto estudios epidemiológicos como clínicos han mostrado una asociación entre la homocisteína y la incidencia de arteriosclerosis. La homocisteína es un factor de riesgo cardiovascular independiente de otros factores de riesgo cardiovascular84,85.
El déficit en la ingesta dietética de folato resulta en hiperhomocisteinemia que acelera la arteriosclerosis por distintas vías:
- -
causa estrés oxidativo86
- -
tiene efectos proinflamatorios
- -
altera el metabolismo lipídico
- -
altera la función endotelial, promoviendo disfunción endotelial87,88 y alterando la función plaquetar e induciendo trombosis89
- -
estimula la proliferación de CMLV.
La relación entre la homocisteína y el riesgo cardiovascular también implica mecanismos de control epigenéticos.
Dos estudios en mujeres posmenopáusicas que recibían una dieta baja en folato mostraron que tenían bajos niveles circulantes de folato, altos niveles de homocisteína y un descenso en el grado de metilación global del ADN en linfocitos90 y en leucocitos91.
El polimorfismo 677C>T en el gen MTHFR (rs1801133) se asocia a una incapacidad de sintetizar 5-metil-HF y por ello se asocia a niveles altos de homocisteína y hipometilación global del ADN92,93.
En monocitos cultivados, cambios en la concentración de homocisteína aumentan los niveles de metilación en ABCA1 y disminuyen los niveles de metilación en ACAT1. Por ello, la metilación del ADN de ABCA1 y de ACAT1 inducida por la homocisteína posiblemente juega un papel importante en la expresión de ABCA1 y ACAT1 y en la acumulación de colesterol en las células espumosas94.
En monocitos también se ha descrito que la hiperhomocisteinemia induce una metilación en la superóxido dismutasa extracelular (EC-SOD), así como aumento en la acetilación de la histona 3 y la histona 495.
La ratio SAM/SAH define el potencial de metilación de una célula. La hiperhomocisteinemia disminuye esta ratio y hace disminuir el potencial metilador.
La epigenética en la arteriosclerosis: presente y futuroLa epigenética ha tenido una trayectoria interesante en el contexto de la biología y la biomedicina. Hace solo 2 décadas, la epigenética era una disciplina exclusiva de pocos biólogos del desarrollo embrionario que buscaban un mecanismo molecular para explicar el enigma de la impronta genómica, es decir el fenómeno por lo cual un alelo está expresado o no dependiendo solamente del sexo del progenitor del cual proviene96. Esos estudios han plasmado una definición inicial de la epigenética como el estudio de mecanismos de regulación transcripcional independiente de la secuencia del ADN. Además, el hallazgo inicial de que los mecanismos de impronta genómica (expresión exclusivamente paterna) en el gen IGF2 están atenuados en tumores de Wilms97 marcó el punto de partida de la epigenética en la biología del cáncer, seguramente la enfermedad en la cual los mecanismos epigenéticos han sido estudiados de manera más intensa y sistemática. En el área cardiovascular, 2 hallazgos fundamentales en años recientes han contribuido a despertar un creciente interés en la epigenética. Primero, el grupo de Jirtle demostró que la dieta puede modificar el epigenoma y la expresión génica de manera estable98. Segundo, el trabajo de Tycko et al. descubrió que por lo menos parte de la información epigenética es alelo-dependiente, lo que implica una interacción entre la información genética y la epigenética99. Estos descubrimientos condujeron a suponer, por un lado, que los factores de riesgo cardiovascular como la dieta, el estilo de vida y otros pueden actuar imponiendo un epigenoma anormal que, a su vez, resulta en un patrón transcripcional aterogénico. Por otro lado, justificaron la idea de que la «missing heritability», evidenciada en las enfermedades cardiovasculares por los estudios de asociación genética, podría ser explicada por la herencia de patrones epigenéticos o por una interacción entre variantes genéticas y epigenoma100. La literatura científica reciente de epigenética cardiovascular parte de estas hipótesis, específicamente, los esfuerzos para describir el epigenoma de la lesión aterosclerótica y por controlar la progresión del ateroma a través de la administración de modificadores epigenéticos. Hasta la fecha, trabajos independientes han demostrado de manera convincente que el ADN de la pared vascular aterosclerótica está hipermetilado y que la inhibición bioquímica de las ADN metiltransferasas disminuye el tamaño del ateroma12,101,102. Indudablemente, estos hallazgos justifican la búsqueda de estrategias clínicamente viables para mantener o revertir la metilación del ADN del tejido vascular aterosclerótico a niveles normales. Los retos inmediatos a los que cualquier proyecto de terapia epigenética cardiovascular se enfrenta son la especificidad del tratamiento y la identificación de un número limitado de blancos genómicos que permitan el eficiente control de la expresión de genes críticos. Una complicación en este aspecto es que la mayoría de la hipermetilación del ADN del ateroma se encuentra en el interior de los genes («gene body») y que poco se sabe sobre la regulación transcripcional por parte de la metilación intragénica12. Sin duda, el progreso de los estudios básicos de epigenética será fundamental para lograr terapias epigenéticas cardiovasculares exitosas. Igualmente importantes han sido los estudios recientes de Epigenome-Wide Association Studies (EWAS), que pretenden identificar asociaciones entre factores de riesgo y patrones epigenéticos en muestras de gran tamaño. Estos importantes estudios identificaron un pequeño número de sitios del genoma cuyo estado de metilación está significativamente asociado con la variación de la insulinemia, VLDL/triglicéridos e IMC103–105. Sin embargo, el efecto de la variación de la metilación sobre dichos parámetros es relativamente modesto. Si son confirmados por otros estudios, estos resultados subrayan la complejidad del reto de identificar un pequeño número de blancos genómicos adecuados para la intervención epigenética. Sin duda, el progreso de la investigación en epigenética básica, en tecnologías de edición del epigenoma y en la descripción del epigenoma vascular revelará si la epigenética podrá tener un papel importante en la clínica cardiovascular.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónEste trabajo se ha llevado a cabo gracias a un proyecto financiado por las Becas FEA/SEA 2011 “Manuel de Oya” para Investigación en nutrición; dos proyectos subvencionados por el Fondo de Investigación Sanitaria FIS (PI10/02547 y PI12/01766); y el CIBERDEM.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.