





El tumor mesenquimal fosfatúrico (TMF) es una causa poco frecuente de osteomalacia oncogénica (OO), síndrome paraneoplásico que puede cursar con clínica severa en el aparato locomotor. El TMF es una neoplasia mesenquimal productora del factor de crecimiento fibroblástico FGF23, a consecuencia del cual se produce hiperfosfaturia e hipofosfatemia resultando en el cuadro de OO. Nuestro objetivo es presentar nuestra experiencia y las complicaciones en el diagnóstico y tratamiento del TMF mostrando una serie de 3 casos.
Material y métodosPlanteamos un estudio observacional, descriptivo y retrospectivo de 3 casos de TMF como causa de OO encontrados tras búsqueda en el sistema de registro digital del hospital y nuestra base de datos de tumores del aparato locomotor. Los criterios de inclusión fueron los siguientes: cuadro clínico de OO, presencia de hipofosfatemia hiperfosfatúrica, niveles elevados de FGF23 en sangre y diagnóstico patológico de TMF.
ResultadosEn todos los casos, la enfermedad comenzo en forma de astenia, dolores óseos inespecíficos, limitación funcional progresiva y presencia de fracturas patológicas. El tiempo medio de retraso diagnóstico fue de 7 años. Analíticamente, todos presentaban una hipofosfatemia hiperfosfatúrica, elevación de los niveles de fosfatasa alcalina y de FGF23. La utilización del octreoscan y el PET/TAC fueron fundamentales para la localización del tumor productor y su posterior biopsia. El tratamiento fue la cirugía en 2 casos y un caso mediante crioterapia dirigida por TAC con control neurofisiológico. Una vez intervenidos se normalizaron los parámetros analíticos. No hemos registrado recidivas hasta la fecha.
ConclusionesEl TMF constituye una entidad rara en el campo de los tumores de partes blandas y óseos, ya que puede darse en ambos tipos de tejido. Debido a la producción del FGF23, ocasiona una alteración en la homeostasis fósforo-calcio. El retraso diagnóstico es la norma, lo cual conlleva a comorbilidades renales y esqueléticas. Para evitarlo, es preciso el conocimiento de la entidad junto con una alta sospecha diagnóstica. El tratamiento quirúrgico conlleva la normalización analítica y del cuadro sistémico.
The phosphaturic mesenchymal tumour (PMT) is a very uncommon cause of oncogenic osteomalacia (OO), which is a paraneoplastic syndrome with severe clinical osteomalacia. The PMT is a neoplasia that produces the fibroblast growth factor FGF23, resulting in reduced proximal tubular phosphate reabsorption leading to hyperphosphaturia and hypophosphatemia. Our aim is to present our experience and complications in diagnosis and treatment of PMT in three patients.
Material and methodsWe propose an observational, descriptive and retrospective study of three cases of OO secondary to PMT found in our database of bone and soft tissue tumours. The inclusion criteria were: symptoms related with OO, presence of hyperphosphaturic hypophosphatemia, elevated levels of FGF23 in blood and pathological diagnosis of PMT.
ResultsIn all cases, the disease showed asthenia, non-specific bone pain, progressive functional weakness, and pathological fractures. The average delay time in diagnosis was 7 years. All presented with hyperphosphaturic hypophosphatemia, elevated levels of alkaline phosphatase as well as FGF23. The use of Octreoscan and PET-CT were essential to find the producing tumour and its subsequent biopsy. Treatment was surgery in two cases and one case was treated by CT-guided cryotherapy with neurophysiological control. Once the surgery was performed, the blood parameters normalized. There is no recurrence.
ConclusionsPhosphaturic mesenchymal tumor is a very rare entity as part of bone and soft tissue tumors, it may occur in both tissues. The phosphate-calcium homeostasis is altered due to high serum levels of FGF23 because of PMT. Delay in diagnosis is usual, leading to renal and skeletal comorbidities. To avoid this, knowledge of this entity together with high diagnostic suspicion are critical. Surgical treatment leads to normalization of serum levels and systemic symptoms.
El tumor mesenquimal fosfatúrico (TMF) es una causa de osteomalacia oncogénica (OO), síndrome paraneoplásico que puede cursar con osteomalacia clínica severa en forma de dolores óseos intensos y fracturas múltiples1. El TMF produce la elevación en sangre del FGF232, hormona sintetizada por esta lesión de origen mesenquimal3,4, en general benigna, de localización anatómica muy variable, y que puede localizarse tanto en el hueso como en las partes blandas5. Epidemiológicamente estas lesiones aparecen a cualquier edad, con un pico de incidencia en la quinta y sexta décadas de la vida6. Su diagnóstico es generalmente tardío, con demoras descritas en la literatura de 5 y 6 años, por su baja prevalencia, sintomatología larvada y poco específica7. La elevación del FGF23 ocasiona hipofosforemia mantenida en relación con la acción fosfatúrica a nivel renal de dicha hormona8. Su diagnóstico se basa fundamentalmente en la detección de niveles elevados de FGF23 en sangre9, así como su demostración con pruebas de imagen como el octreoscan o el PET/TAC con 25-fluorodesoxiglucosa10. El tratamiento de la OO secundaria al TMF, se basa en la localización y en la resección del tumor responsable y productor del FGF2311,12.
Nuestro objetivo es presentar nuestra experiencia y complicaciones en el diagnóstico y tratamiento del TMF, a propósito de 3 casos tratados en nuestro centro, poniendo el acento en la importancia de la valoración del metabolismo del fósforo y calcio, y la alta sospecha diagnóstica.
Material y métodosSe trata de un estudio retrospectivo y análisis de la evolución en una serie de tres pacientes, identificados tras la búsqueda exhaustiva en nuestra base de oncología ortopédica, así como en la herramienta Excalibur® de nuestro hospital (identifica cualquier palabra escrita en informes de alta o quirúrgicos desde 1978) utilizando los términos «tumor mesenquimal fosfatúrico» «osteomalacia oncogénica» e «hipofosforemia». Los criterios de inclusión son los considerados como diagnósticos de esta entidad: cuadro clínico compatible con osteomalacia, presencia analítica de hipofosforemia (fósforo >4,5mg/dl) con hiperfosfaturia en orina de 24h (>950mg), niveles elevados de FGF23 en sangre (>3,5U/I), determinación de su existencia mediante pruebas de imagen bien de radiología convencional (radiografía, TAC y RMN) o de medicina nuclear (octreoscan, gammagrafía ósea y PET/TAC con 25-fluorodesoxiglucosa) y diagnóstico patológico de TMF (usando tinciones de hematoxilina-eosina). Se incluyeron datos epidemiológicos (edad, sexo), anamnesis (año de inicio de síntomas, motivo de derivación a cirugía ortopédica), exploración clínica (presencia de fracturas patológicas, localización de las mismas), pruebas de radiodiagnóstico (radiografía, TAC o RMN), pruebas de medicina nuclear (octreoscan o PET/TAC con 25-fluorodesoxiglucosa), datos de laboratorio con niveles de fósforo, calcio, fosfatasa alcalina (FA), hormona paratiroidea (PTH), calcitriol y FGF23.
El seguimiento medio de los pacientes ha sido de 52 meses (rango: 48-60 meses).
ResultadosA continuación, ofrecemos una descripción de los 3 casos (datos más relevantes resumidos en las tablas 1-4).
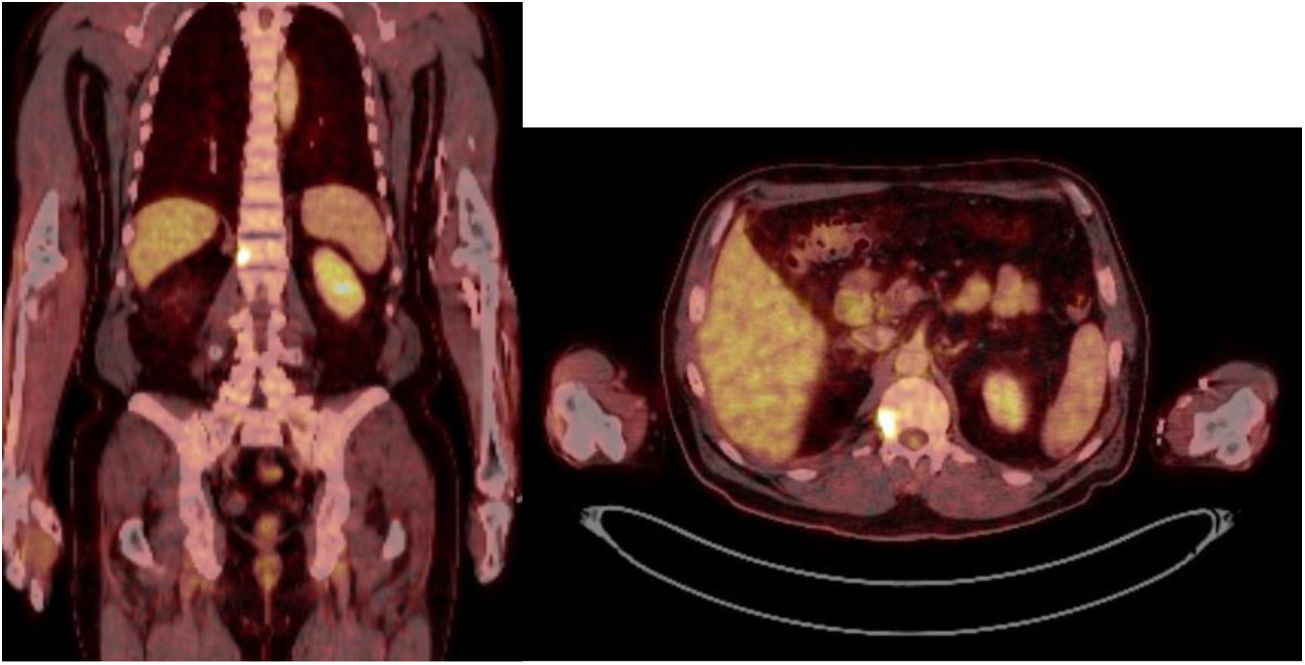
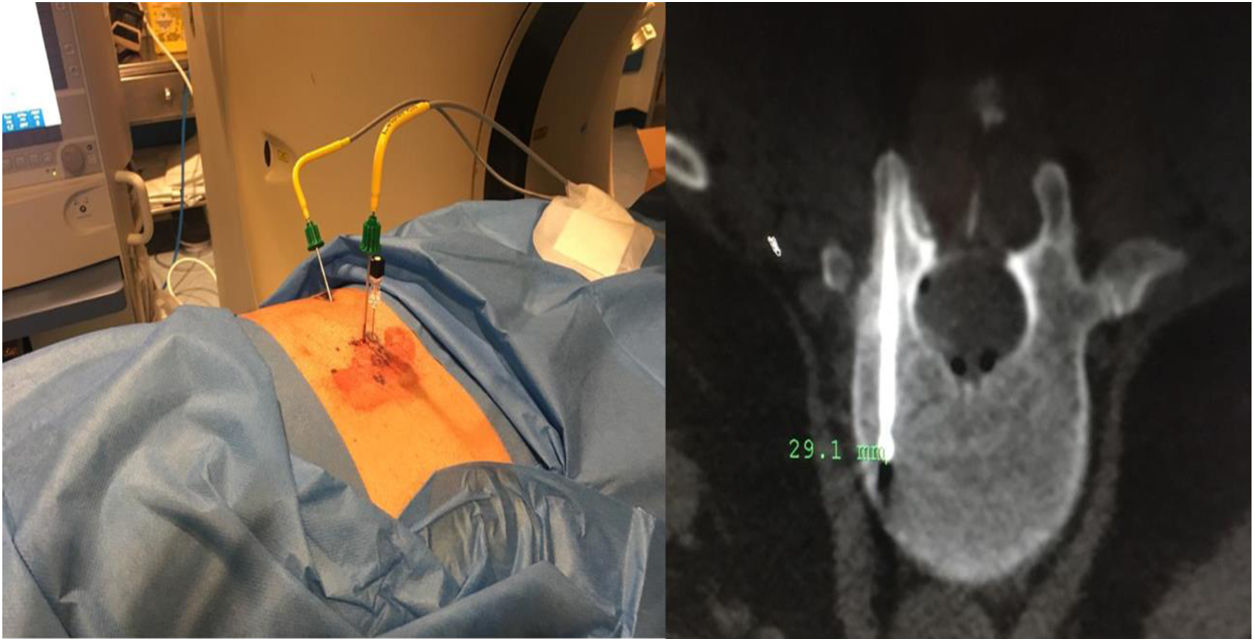
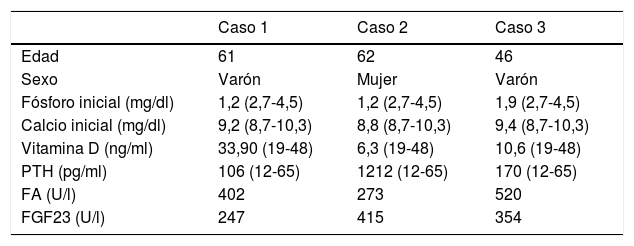
Caso 1Varón de 61 años, sin antecedentes de interés, valorado por hipofosfatemia severa e hiperfosfaturia refractaria a tratamiento. Clínicamente, dolor óseo general incapacitante, precisando muletas para caminar. Las imágenes mostraron lesiones óseas múltiples, y fracturas a varios niveles, destacando fractura subcapital de fémur derecho no desplazada. La PET/TAC detectó nódulos tiroideos bilaterales con PAAF negativa. La gammagrafía marcada con Tc-sestamibi presentó captación focal compatible con adenoma paratiroideo, orientando el diagnóstico hacia un hiperparatiroidismo primario (HPTP). Se practicó una hemitiroidectomía izquierda resultando en glándula paratiroidea normal. El nuevo estudio analítico resultó en hipofosforemia de 1,2mg/dl, FA de 402U/l y FGF23 de 247U/l (n<145). Dicho hallazgo enfocó el diagnóstico hacia el TMF, no detectable inicialmente con RMN y PET/TAC. Así, se optó por tratamiento médico sustitutivo, mejorando el paciente de forma parcial a nivel clínico y analítico. Debido a los niveles de FGF23 hasta 357,2U/l se realizó un octreoscan sin diagnóstico. Finalmente, la repetición del PET/TAC detectó aumento de actividad metabólica en el hemicuerpo derecho de L1 (fig. 1). Tras biopsia con aguja gruesa (BAG) guiada por TAC se diagnosticó lesión compatible con TMF. El tratamiento consistió en crioterapia percutánea de la lesión guiada por TAC y control neurofisiológico simultáneo (fig. 2). Durante el evolutivo se han normalizado los niveles de fósforo en sangre y orina y de FGF23.


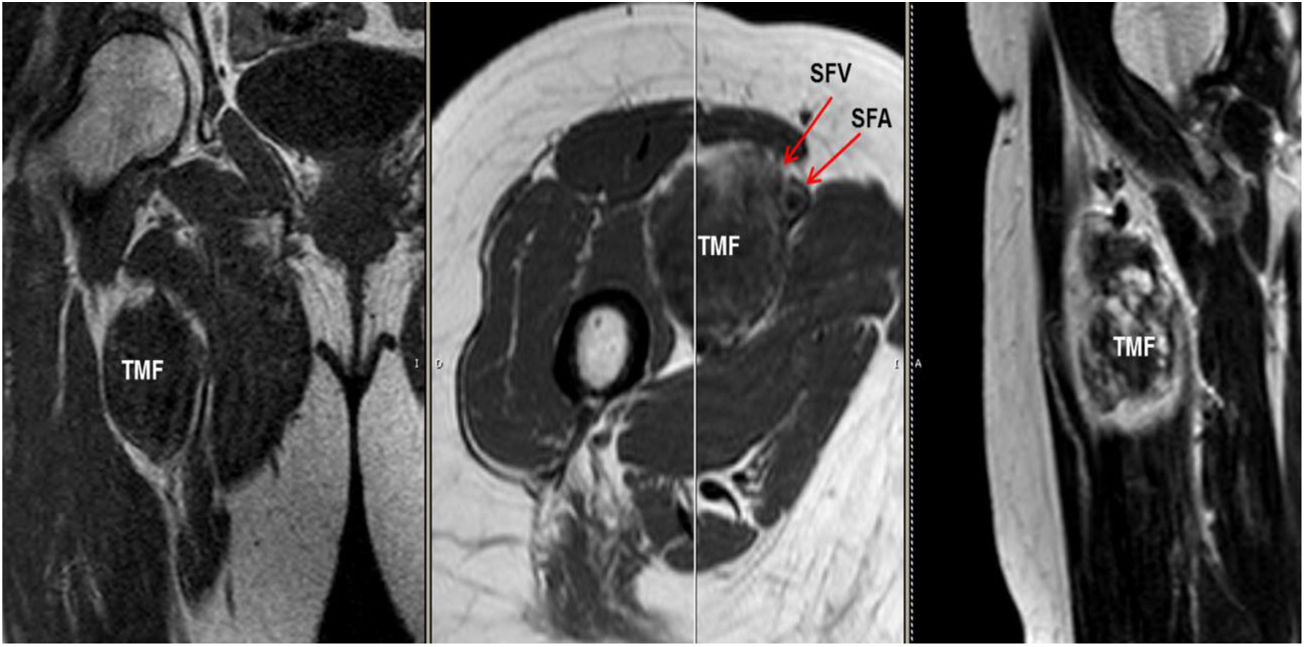
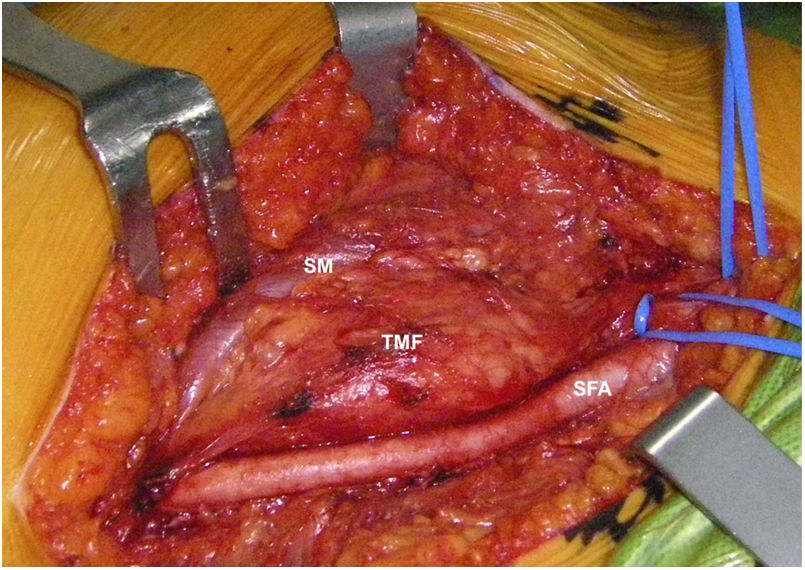
Mujer de 62 años, con antecedentes de hemitiroidectomía izquierda por hiperplasia nodular. Clínicamente comenzó con dolor óseo generalizado de 2 años de evolución, tratado con AINE. Posteriormente, se objetivó hipofosfatemia e hiperfosfaturia, FA de 273U/l, osteopenia radiológica severa con fracturas de las vértebras T11-T12-L1-L2, sin traumatismo. Inicialmente, se diagnosticó de osteomalacia hipofosforémica del adulto, comenzando tratamiento médico con fósforo y calcitriol, seguido de mejoría clínica. A continuación, inició deterioro progresivo de la función renal con hipofosfatemia mantenida a pesar del tratamiento médico. Ello condujo a enfermedad renal crónica estadio IV con nefrocalcinosis cortical bilateral. Consecutivamente, se detectó elevación de la PTH a 1.212pg/ml, FGF23 de 415U/l (n<145), creatinina de 2,72mg/dl y fósforo de 1,7mg/dl. Con estos resultados, el octreoscan detectó captación en ingle derecha, con masa de partes blandas a ese nivel. Tras la RMN (fig. 3) se realizó BAG guiada por ecografía, resultando en TMF. Se realizó resección amplia (fig. 4) con confirmación histológica de TMF, y normalización inmediata analítica. Desafortunadamente, esta paciente presentó, dentro de las complicaciones crónicas de los raquitismos hipofosforémicos, el HPTS y la nefrocalcinosis.

.")
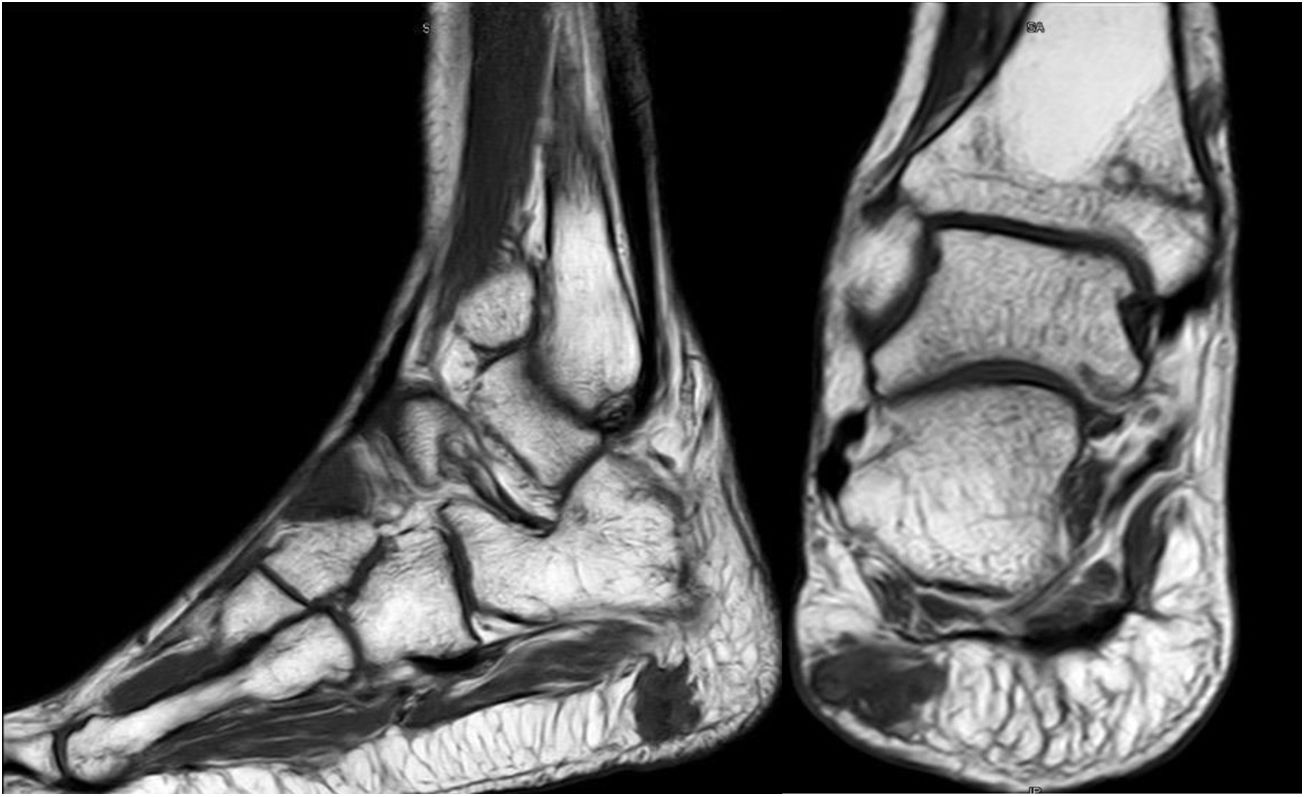
Varón de 46 años, sin antecedentes de interés, que en 2012 comienza con dolor lumbar irradiado tras esfuerzo. En la RMN inicial presentó hernia discal central L5-S1 y hernia lateral y foraminal izquierda L3-L4. Sin mejora clínica con tratamiento conservador, se optó por descompresión y artrodesis instrumentada en los niveles L3-S1 en septiembre de 2013. Clínicamente empeoró, mostrando dificultad para la deambulación, precisaba de 2 muletas, y finalmente silla de ruedas. Durante el seguimiento observamos seudoartrosis en los niveles intervenidos y aflojamiento aséptico de la instrumentación. Se reintervino en marzo de 2014, retirando el material previo, colocando uno nuevo y añadiendo injerto. La evolución fue desfavorable, precisando una tercera cirugía en junio de 2015, añadiéndose por vía anterior cajas intersomáticas con nuevo injerto autólogo. La falta de mejoría motivó un estudio del metabolismo óseo que evidenció hipofosfatemia hiperfosfatúrica y elevación de FGF23. Además, se produjo una fractura de cuello del fémur izquierdo. El octreoscan halló un aumento de actividad en la almohadilla plantar del talón derecho. La RMN confirmó la lesión de partes blandas (fig. 5). Finalmente, dado el pequeño tamaño de la lesión y la alta sospecha diagnóstica, se realizó resección amplia, confirmándose TMF. El paciente evolucionó de forma favorable, con normalización clínica y analítica, siendo independiente para las actividades de la vida diaria. Desafortunadamente, ha precisado de prótesis total de cadera bilateral debido a osteonecrosis a este nivel.

Resumen en las tablas 1-4 de los principales datos de los 3 casos clínicos presentados.
DiscusiónLa hipofosfatemia es una alteración relativamente común, descrita hasta en un 5% del total de los pacientes hospitalizados13, cuyo déficit prolongado da lugar a alteraciones musculares y trastornos óseos. De este modo, cuando la situación clínica presenta un origen tumoral, puede desarrollarse un cuadro de OO, que, si es severa y se prolonga en el tiempo, puede contribuir a una gran morbilidad renal y del aparato locomotor14.
En los 3 casos expuestos, la causa de OO ha sido el TMF, el cual, puede presentarse a cualquier edad, siendo más frecuente su aparición entre la quinta y la sexta década de vida (tabla 1). Su diagnóstico es generalmente tardío, con retrasos reportados en 5 o 6 años6,7, que, en nuestros casos, resultó en un retraso medio de 80 meses (tabla 4). El primero en reportar la existencia de esta entidad fue McCance15 en 1947, pero fue Prader et al.16 en 1959, quienes reconocieron el tumor como posible causa de osteomalacia. La mayor parte de estas lesiones suelen asentar sobre tejidos blandos o, en menor medida, en huesos a nivel de extremidades inferiores, tronco, cabeza y cuello. Microscópicamente2 están compuestos por células pequeñas, redondeadas, fusiformes o en forma de huso dentro de una matriz difusa que contiene un delicado patrón de capilares. La matriz tiende a calcificarse dando lugar a depósitos de mineral situados entre las células.
Datos epidemiológicos y de laboratorio
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| Edad | 61 | 62 | 46 |
| Sexo | Varón | Mujer | Varón |
| Fósforo inicial (mg/dl) | 1,2 (2,7-4,5) | 1,2 (2,7-4,5) | 1,9 (2,7-4,5) |
| Calcio inicial (mg/dl) | 9,2 (8,7-10,3) | 8,8 (8,7-10,3) | 9,4 (8,7-10,3) |
| Vitamina D (ng/ml) | 33,90 (19-48) | 6,3 (19-48) | 10,6 (19-48) |
| PTH (pg/ml) | 106 (12-65) | 1212 (12-65) | 170 (12-65) |
| FA (U/l) | 402 | 273 | 520 |
| FGF23 (U/l) | 247 | 415 | 354 |
FA: fosfatasa alcalina; FGF23: Factor de crecimiento fibroblástico 23; PTH: hormona paratiroidea.
El diagnóstico del síndrome de OO por TMF, aunque sencillo, precisa de una alta sospecha diagnóstica17,18. Observamos una osteomalacia severa, con hipofosforemia significativa causada por una fosfaturia inapropiadamente elevada (por tanto, con una baja tasa de reabsorción tubular de fosfatos [RTP]), calcio y PTH normales (al menos inicialmente), niveles de calcitriol bajos o inapropiadamente normales para la hipofosforemia, FA aumentada significativamente y resistencia al tratamiento con vitamina D19 (tabla 1). Estos hechos son diferenciales respecto del hiperparatiroidismo primario (HPTP), que generalmente cursa con hipercalcemia, hipofosfatemia y niveles séricos elevados de PTH. Al contrario, el HPTS por déficit de vitamina D cursa con niveles de calcio normales. Además, en el HPTS a insuficiencia renal crónica hallamos hipocalcemia, hiperfosforemia, niveles elevados de PTH y bajos de calcitriol20. Posteriormente, el diagnóstico sindrómico se confirma con un valor de FGF23 sérico muy elevado9 (tabla 1). Cuando el cuadro aparece en edades tempranas de la vida, debe hacerse el diagnóstico diferencial con los raquitismos hipofosforémicos, para lo cual es de relevancia la presencia o no de familiares con un síndrome semejante, y la evidencia o no de un tumor mesenquimal21. Una elevación de FGF23 con hipofosforemia e hipofosfaturia se produce también en el síndrome de McCune-Albright22 y en el síndrome de los nevus epidermicos23, pero en ambos casos las lesiones cutáneas y otras manifestaciones son evidentes desde el primer momento. Por otro lado, la recaída de la enfermedad parece relacionada con la elevación en los niveles de FGF23 en sangre y fósforo en orina24.
En nuestros pacientes (tabla 1), observamos una hipofosfatemia importante con valores en suero en torno a 1,2-1,9mg/dl, acompañada de un cuadro de osteomalacia caracterizado sistémicamente por astenia importante, así como una clínica esquelética consistente en fracturas patológicas o necrosis avascular femoral (tabla 2), que condicionaron una impotencia funcional para la marcha. De forma independiente, las complicaciones crónicas derivadas de los raquitismos hipofosforémicos tales como el hiperparatiroidismo o la nefrocalcinosis19 aparecieron en uno de nuestros casos (tabla 2).
Datos clínicos de afectación esquelética, sistémica y secuelas
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
| Localización de las fracturas patológicas | Fémur proximal derecho | Vértebras T11-T12-L1-L2 | Basicervicales ambos fémures y aplastamientos vertebrales |
| Clínica sistémica | Debilidad generalizada | Debilidad generalizada, astenia | Impotencia funcional progresiva |
| Secuelas | No | Insuficiencia renal crónica e hiperparatiroidismo terciario | Artroplastia bilateral de cadera por osteonecrosis |
Característicamente, como se ha comprobado en nuestros casos, esta osteomalacia es resistente al tratamiento con vitamina D y, en tanto en cuanto no se detecte y suplemente la hipofosfatemia, no se corrige la sintomatología6. En los sujetos de nuestra serie, el tratamiento médico de la hipofosfatemia tardó en concretarse. Tras la instauración del mismo, se obtuvo una lenta, aunque paulatina recuperación de la osteomalacia y de la morbilidad concomitante, pero, ante el mantenimiento de la hiperfosfaturia, acabó con la persistencia del cuadro clínico. Aun así, el tratamiento sintomático no diagnostica ni trata la causa del problema, el cual radica en identificar los niveles séricos altos de FGF23 y la localización anatómica de la neoplasia productora del mismo25. La determinación del FGF23 mediante técnicas de ELISA se estima que puede confirmar el diagnóstico clínico con una sensibilidad estimada del 23-86% estando el tumor sin identificar, y del 38-100% con tumor confirmado24. Sin embargo, se han descrito casos de TMF con FGF23 normal26. En todos nuestros casos, una vez se tuvo la sospecha de TMF, dentro del diagnóstico diferencial de raquitismo hipofosfatémico, se observaron elevaciones muy importantes del FGF23 (tabla 1).
La localización anatómica de la neoplasia productora radica en un examen físico detallado, acompañado de las pruebas de imagen: octreoscan, PET/TAC con 25-fluorodesoxiglucosa y RMN10. Además, en los últimos años se han incorporado nuevas pruebas de imagen como el Ga-DOTANOC PET/TAC (utilizando una molécula de octeótrido modificada) o el muestreo venoso con FGF23 en zonas donde los estudios de imagen sugieren lesiones sospechosas25,10. En nuestro centro se optó por el uso del octreoscan y PET/TAC con 25-fluorodesoxiglucosa para la localización anatómica de la neoplasia, y después la RMN para identificación y planificación de biopsia y tratamiento. Aun así, debido al tamaño que en ocasiones presenta el TMF, no es posible detectarlo inicialmente por estos métodos, por lo que se recomienda la repetición de las pruebas de imagen durante el seguimiento27, hecho también advertido en nuestros casos (tabla 3).
Datos de imagen
| Caso 1 | Caso 2 | Caso 3 | |
|---|---|---|---|
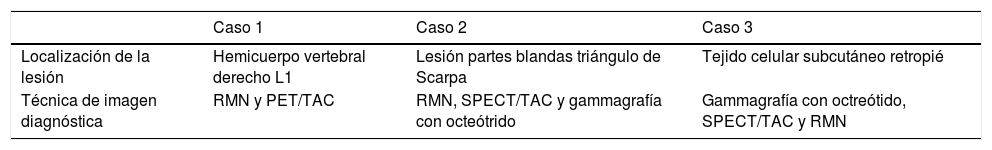
| Localización de la lesión | Hemicuerpo vertebral derecho L1 | Lesión partes blandas triángulo de Scarpa | Tejido celular subcutáneo retropié |
| Técnica de imagen diagnóstica | RMN y PET/TAC | RMN, SPECT/TAC y gammagrafía con octeótrido | Gammagrafía con octreótido, SPECT/TAC y RMN |
PET/TAC: tomografía por emisión de positrones/tomografía axial computarizada; RMN: resonancia magnética nuclear; SPECT/TAC: tomografía computarizada por emisión de fotón único.
El tratamiento definitivo del TMF consiste en su resección quirúrgica28, lo cual va seguido de una rápida mejoría analítica y la consiguiente regresión de los síntomas. Los casos 2 y 3 fueron tratados quirúrgicamente mediante resección amplia. El caso 1 se optó por ablación percutánea con crioterapia guiada por TAC, bajo control neurofisiológico. Esta técnica está ampliamente difundida para el tratamiento percutáneo de lesiones benignas de origen mesenquimal, incluyendo el propio TMF29-32. Basado en la experiencia de nuestro grupo, creemos que esta modalidad terapéutica aporta grandes ventajas no solo en la erradicación de la lesión, sino también en la minimización de complicaciones y recuperación de los pacientes en localizaciones anatómicas complejas.
En la literatura, se ha descrito algún caso de TMF con signos histológicos de malignidad y desarrollo de metástasis, pero este hecho es muy poco frecuente33-35 y no es nuestra experiencia. Debido a ello, y a la posible reaparición de los síntomas por recaída local de la lesión, el seguimiento en estos pacientes ha de ser a largo plazo. Además, tras el tratamiento, el pronóstico funcional es generalmente bueno, salvo en aquellas complicaciones severas renales o a nivel del esqueleto en las que no es posible su regresión27. Nuestros pacientes han mejorado completamente de la osteomalacia, teniendo que tratar las secuelas de las mismas, fracturas patológicas y osteonecrosis de cadera (tabla 2). Tras la cirugía, todos nuestros pacientes experimentaron normalización de los niveles de FGF23 y fósforo en sangre y orina. Tal y como describe en su artículo Jonsson et al. en 2003, aumentos posquirúrgicos en el valor del FGF23 suele traducirse en recidiva local de la lesión26. En nuestros casos, no se ha presentado hasta la fecha dicha recaída, aunque el seguimiento es corto (tabla 4). En aquellos raros casos en los que no se identifique el tumor productor de FGF23, el tratamiento se basa en la terapia sustitutiva con suplementos de fósforo y calcitriol36, o la posible utilización del burosumab. Este fármaco, anticuerpo monoclonal anti-FGF23, solo está aprobado en la actualidad por la FDA para su uso regular en el tratamiento de la hipofosfatemia ligada al cromosoma X37, aunque ya existen ensayos clínicos específicos en la indicación del FGF23 en el manejo de la osteomalacia oncogénica38,39.
Nuestro estudio presenta grandes limitaciones. Es un estudio retrospectivo, con casuística de 3 casos y un seguimiento, hasta el momento, limitado. La mayoría de las referencias consultadas presentan casuísticas muy limitadas25,40-42, siendo la serie más numerosa recogida hasta la fecha, la publicada por Feng et al. en 2017 con 144 casos27, aunque recogidos en un intervalo de 32 años, resultando en una media aproximada de 4 casos anuales. Respecto al diseño del estudio, resaltar que algunos de los trabajos de mayor impacto revisados2,11,27, presentan un diseño retrospectivo, diseños que se ven favorecidos en contexto de patologías poco frecuentes. En cuanto al tiempo de seguimiento de los pacientes, salvo la excepción del estudio de Feng et al. de 201727 que recoge tiempos de seguimientos superiores, existen artículos publicados con tiempos de seguimiento similares a los nuestros11. En resumen, nuestras observaciones se asemejan a las de las distintas publicaciones previas, en cuanto a demora en el diagnóstico, presentación clínica y analítica, anatomía patológica, y métodos de diagnóstico, tratamiento, evolución y seguimiento. Por tanto, debido a la rareza de esta entidad, creemos que es importante su comunicación dadas las implicaciones clínicas en cuanto a morbilidad que produce el TMF puesto que, desafortunadamente, el retardo diagnóstico es lo habitual.
ConclusiónEl TMF es una entidad nosológica muy poco frecuente, causante de un síndrome de OO hipofosfatémica e hiperfosfatúrica resistente al tratamiento con vitamina D. Su rareza como entidad y su clínica larvada e inespecífica hace posible que pase inadvertido entre las distintas especialidades que abordamos esta patología: cirugía ortopédica, endocrinología, nefrología, radiodiagnóstico y reumatología. Por tanto, es obligado ante la presencia de un cuadro de raquitismo hipofosfatémico, incluir esta entidad en el diagnóstico diferencial, realizando un exhaustivo estudio del metabolismo del fósforo y calcio, obteniendo niveles de FGF23 y realizando pruebas de imagen (octreoscan, el PET/TC y la RMN), con el fin de descartar o identificar la presencia de TMF. La resección del TMF es la primera opción terapéutica. Cuando la lesión se localice en situaciones anatómicas complejas para cirugía o por la morbilidad que esta pudiera ocasionar, el tratamiento percutáneo mediante radiofrecuencia o crioterapia guiado por TC es una opción plausible. Recomendamos que ante la sospecha clínica de esta entidad o en caso de confirmarse se remitan a centros de referencia.
Nivel de evidenciaNivel de evidencia IV.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr. D. Ignacio Sánchez del Campo, pionero a nivel nacional del conocimiento de la enfermedad tumoral musculoesquelética.
