


The use of specific antibodies in inflammatory myopathies has improved the characterization of this disease, identifying different clinical phenotypes. Patients with dermatomyositis (DM) and anti-MDA5 antibodies display typical skin symptoms, lesser muscular involvement, and a prevalence of interstitial lung disease (ILD) of up to 91%. Beyond ILD, spontaneous pneumomediastinum (SN) has been identified as a rare but potentially fatal pulmonary manifestation. Two cases of this complication in patients with anti-MDA5 DM are reported.
El uso de anticuerpos específicos en miopatías inflamatorias ha mejorado la caracterización de esta enfermedad identificando distintos fenotipos clínicos. Los pacientes con dermatomiositis (DM) y anticuerpos anti MDA5 muestran síntomas típicos en piel, menor compromiso muscular y una prevalencia de enfermedad pulmonar intersticial (EPI) de hasta el 91%. Además de la EPI, se ha identificado el neumomediastino espontáneo (NE) como una manifestación pulmonar rara pero potencialmente mortal. Se reportan 2 casos de esta manifestación en pacientes con DM anti MDA5.
Dermatomyositis (DM) was characterised in the literature by its muscle and skin involvement; however, the involvement of other clinical domains, such as the lung, is increasingly recognised.1 Thus, patients with anti-MDA5 antibodies have only mild muscle involvement confirmed by electromyography or a subtle CPK elevation, known as hypomyopathic or amyopathic DM, which is absent in some cases but is associated with a high risk of developing pulmonary involvement during the course of their disease.2
Interstitial lung disease (ILD) clearly tops the list of the major pulmonary manifestations in patients with DM in the presence of anti-MDA5 antibodies, not only because it is the most prevalent, but also because of its high burden of morbidity, even causing death in those patients where it progresses rapidly. However, this is not the only condition affecting the lung in these subjects; other less frequent manifestations have also started to be documented, such as aspiration pneumonia secondary to respiratory muscle weakness, diffuse alveolar damage, and bronchiolitis obliterans.3 Other manifestations have been described much less frequently, such as subcutaneous emphysema, pneumothorax, and spontaneous pneumomediastinum (SP), which, although not very prevalent, should be recognised and diagnosed in a timely manner because they carry a high burden of morbidity and mortality in patients, not only due to the condition per se, but also because they are markers of rapid progression of ILD.4,5 Below we present 2 cases of SP in patients with anti-MDA5 antibodies.
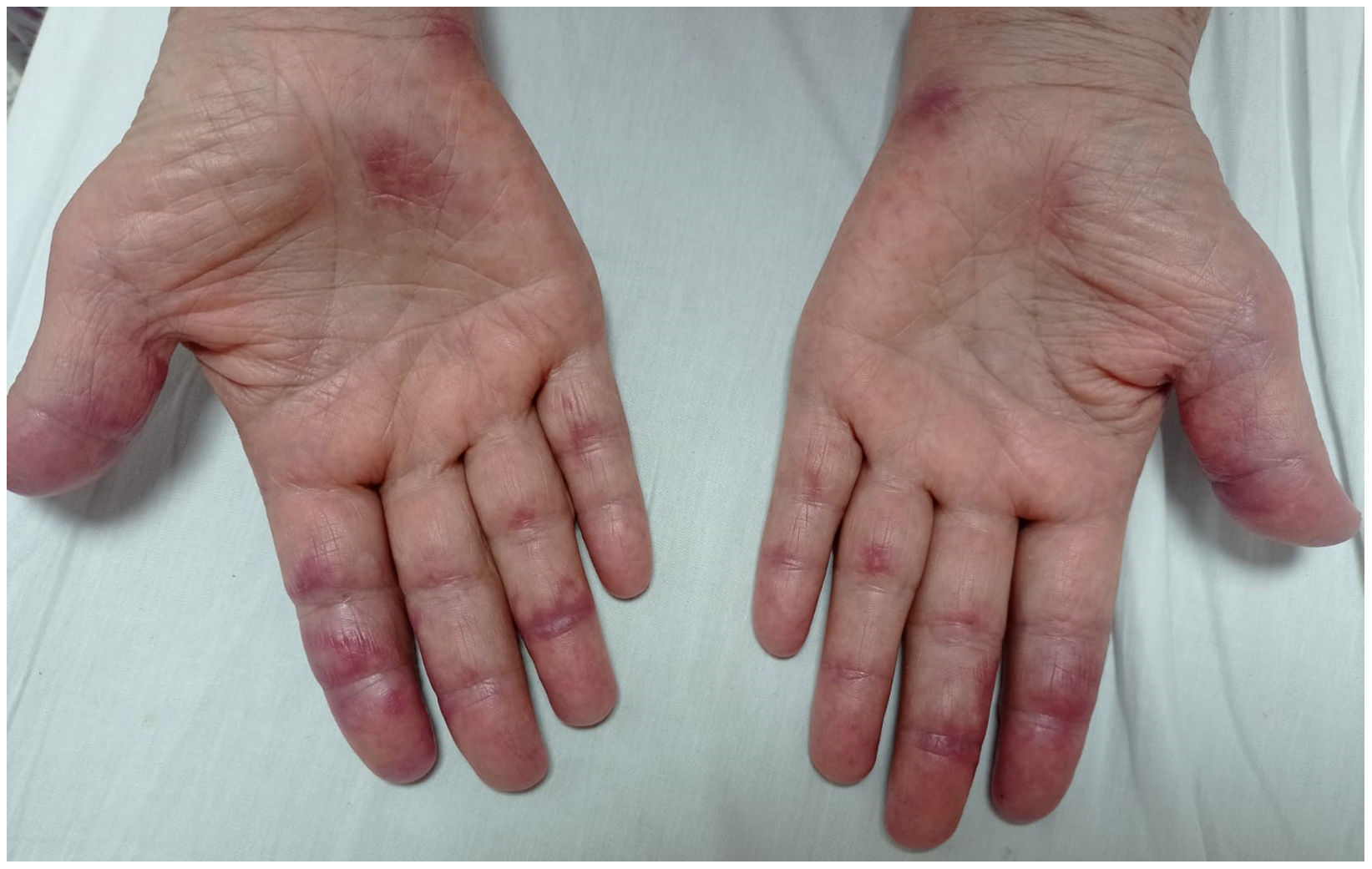
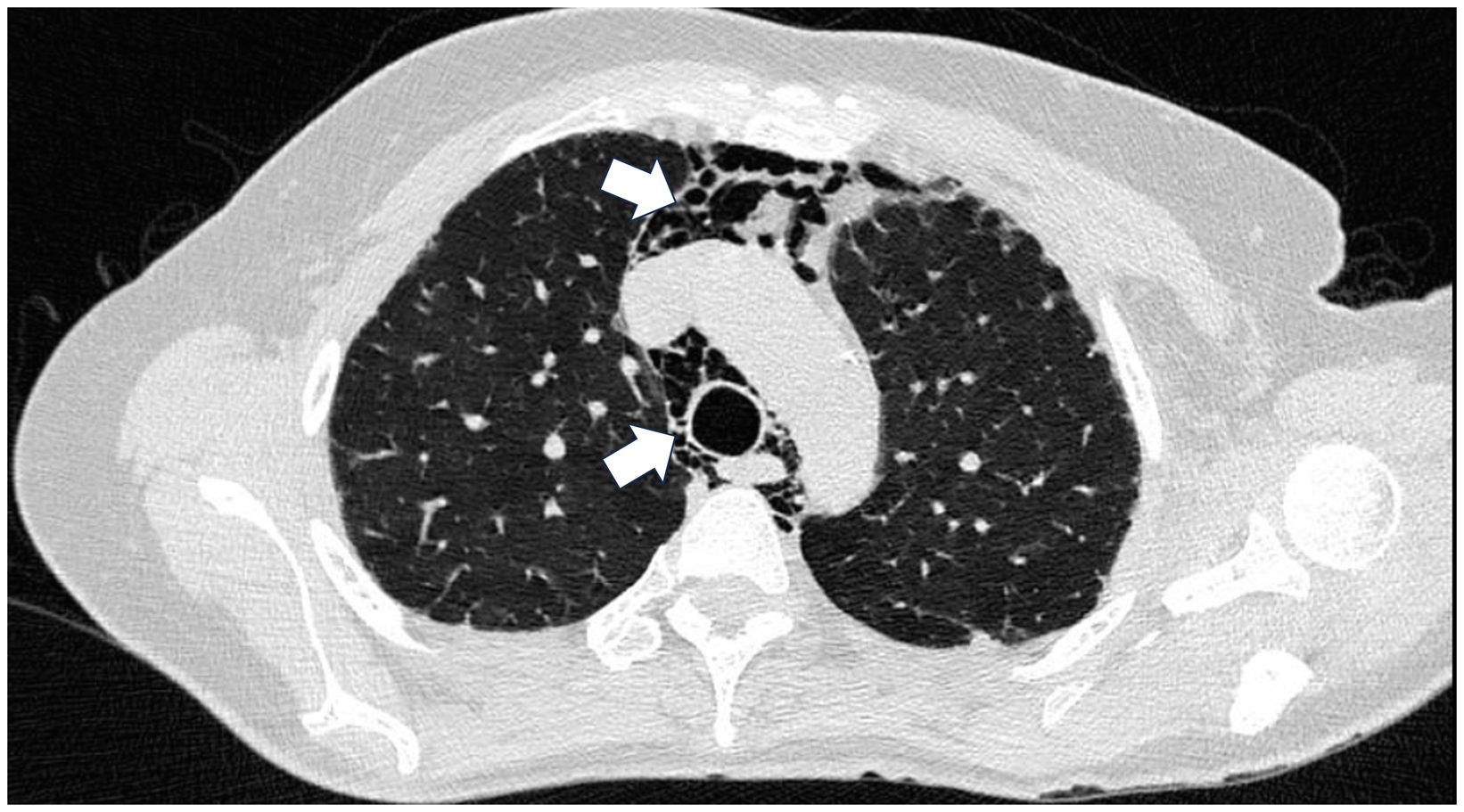
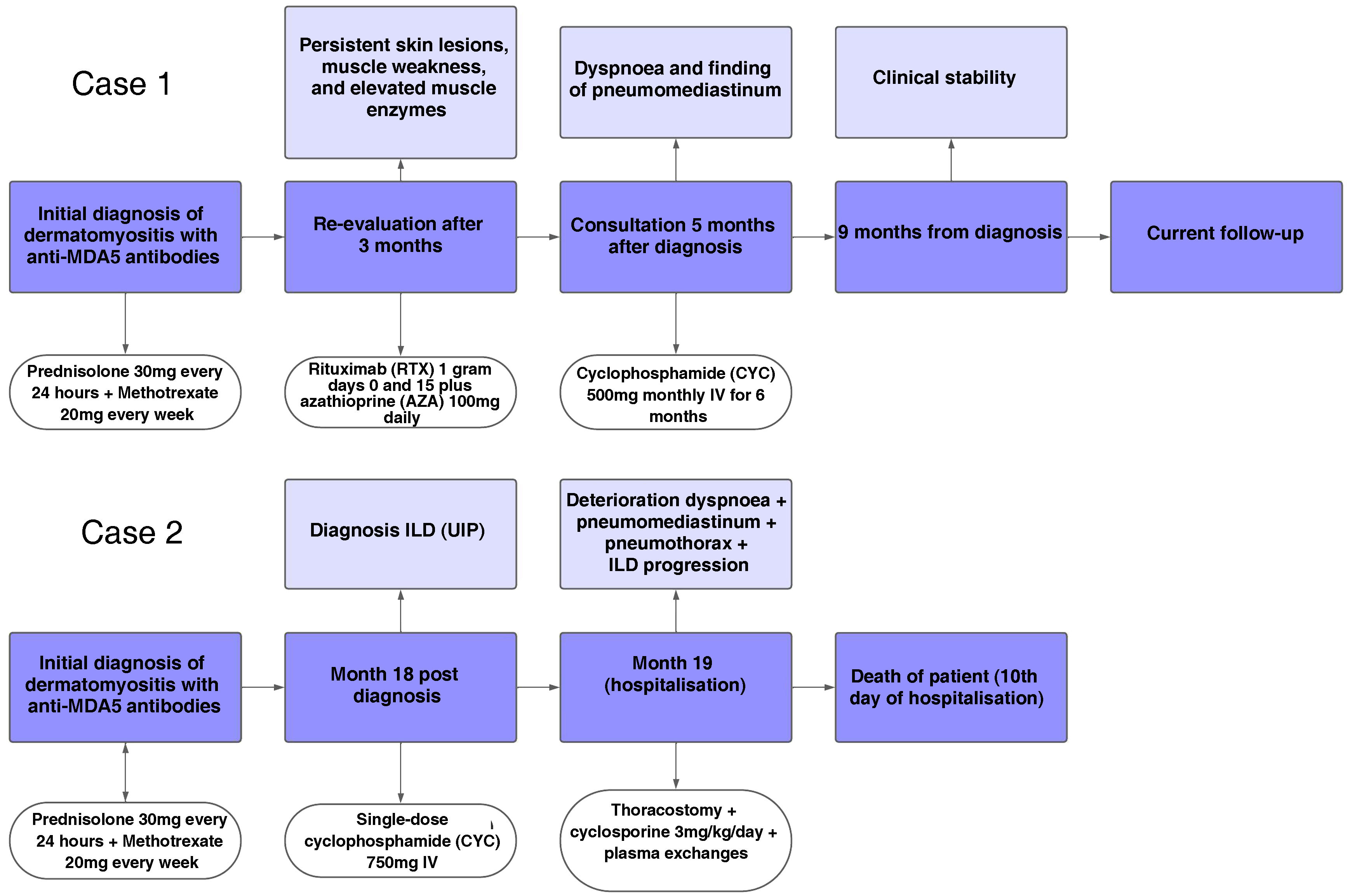
Case presentationCase 1A 60-year-old woman with a history of DM with positive anti-MDA5 antibodies diagnosed one year ago presented at the time of diagnosis with skin involvement of heliotrope rash, shawl sign, Gottron's sign, and violaceous and ulcerated lesions on the palms, polyarthritis, proximal muscle weakness of all 4 limbs, and cephaloparesis. At baseline, she was prescribed methotrexate 20 mg weekly and prednisolone 20 mg daily in a tapering regimen. After 3 months of follow-up, her skin lesions, muscle weakness, and marked elevation of muscle enzymes persisted, and she also had alopecia, a potential adverse effect of methotrexate was therefore considered, in addition to clinical refractoriness, and we decided to start the first cycle of rituximab 1 g IV on days 0 and 15, plus azathioprine 100 mg daily. She consulted after 2 months due to persistence of skin lesions and polyarthritis, despite improvement in muscle weakness and normalisation of muscle enzymes. Physical examination revealed swelling of the proximal interphalangeal, metacarpophalangeal, carpal and elbow joints, with additional heliotrope rash, Gottron papules, macules, and erythematous-violaceous plaques with incipient signs of ulceration (Fig. 1). There were also crypts in both lung fields, and therefore a high-resolution CT scan was requested, which showed isolated extensive pneumomediastinum without the presence of pneumothorax or cutaneous emphysema (Fig. 2). Oesophageal perforation was ruled out by upper gastrointestinal endoscopy and bronchial lesion by bronchoscopy; therefore, this finding was attributed to a manifestation of DM, in the absence of ILD. Given the clinical stability and the absence of respiratory symptoms, cyclophosphamide 500 mg IV monthly for 6 months was started. She is currently being monitored by the outpatient clinic awaiting completion of the proposed regimen, with clinical improvement of the skin lesions and dyspnoea.


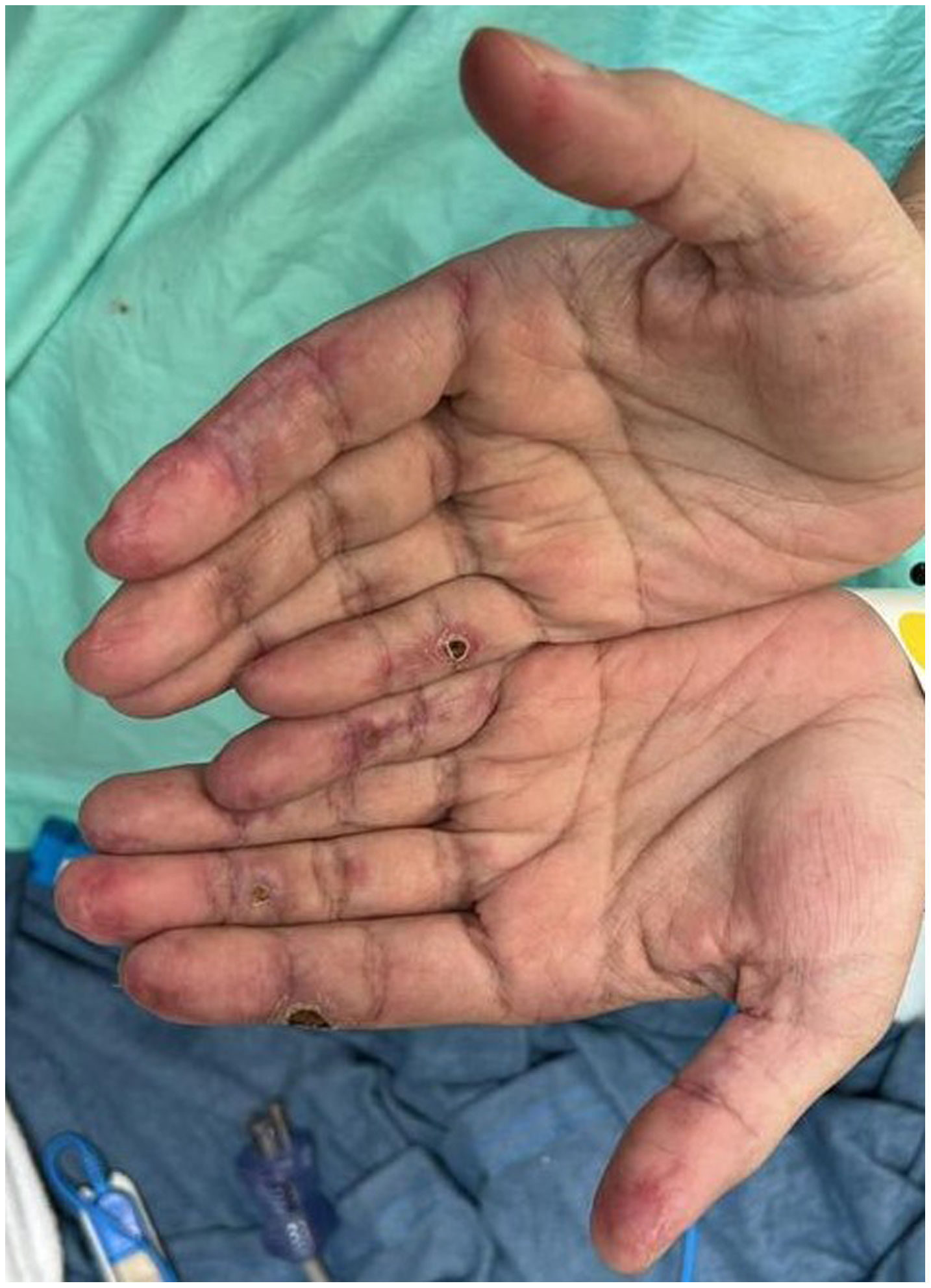
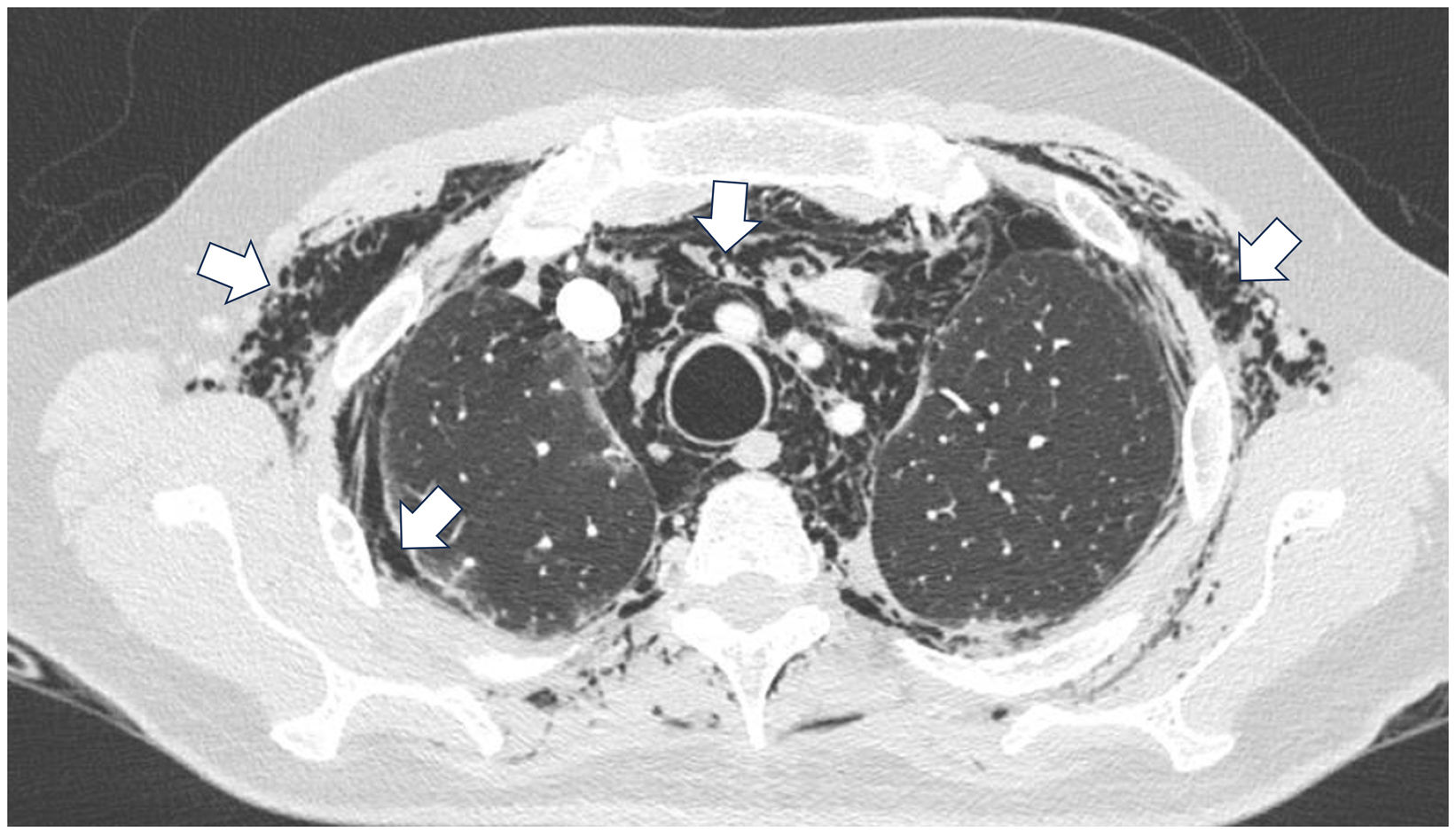
A 45-year-old man with a 2-year history of DM with positive anti-MDA5 antibodies and the classic muscular and cutaneous manifestations of this clinical phenotype (Fig. 3), in addition to ILD of the usual interstitial pneumonia type, which had been diagnosed 2 months earlier and for which he had been treated with a single dose of cyclophosphamide 750 mg IV. He consulted spontaneously due to sudden dyspnoea, and therefore underwent an angio-CT scan (Fig. 4) which showed, in addition to the underlying pulmonary fibrosis, concomitant pneumomediastinum and pneumothorax. Bronchoscopy and microbiological studies were performed, which were negative, and therefore we decided to continue with the next dose of cyclophosphamide, cyclosporine 3 mg/kg/day and start plasma exchanges. However, in the following days, after starting the therapies, the patient showed further respiratory deterioration and unfortunately died due to progression and exacerbation of ILD, in addition to a considerable increase in pneumomediastinum and pneumothorax despite having undergone thoracostomies by thoracic surgeons. The whole picture was considered to constitute a diagnosis of rapidly progressive ILD, and the patient eventually died.


A brief timeline with the respective outcomes in each case is shown in Fig. 5.
Discussion
SP has been described as a rare manifestation in different autoimmune diseases and disorders such as systemic sclerosis, rheumatoid arthritis, and systemic lupus erythematosus. This association has been reported most frequently in DM, with an incidence ranging from 2.2% to 8.6% for this entity alone, with no data reported for other autoimmune diseases.6,7 However, these data are not very consistent, because most of the information found in the literature are case reports and case series. Some authors, such as Civit and Porcel,8 even indicate that fewer than 100 cases had been described by 2020. Despite its low prevalence, recent Chest guidelines on pulmonary disease associated with myositis highlight the importance of this radiological finding because it is associated with worse outcomes.7
Although SP as a complication of DM has been described since 1986,9 the pathophysiological mechanisms causing this condition are still unknown, the main hypothesis being a vasculopathic phenomenon on the respiratory epithelium that leads to alveolar rupture and/or formation of subpleural cysts, with the consequent formation of SP.10 Although many cases have been reported with concomitant ILD, it is not impossible for SP to occur in isolation, as occurred in one of our cases, where there was an absence of ILD, a circumstance that has also been reported in other similar cases, which shows that there is an underlying pathophysiological process other than that which produces ILD, and that the latter is not a prerequisite for the onset of SP.3,11,12
Risk factors described prior to the routine use of myositis-specific antibodies were a history of ILD, amyopathic disease behaviour, and cutaneous vasculopathy,10,13 which are ultimately the most characteristic manifestations of DM associated with anti-MDA5, and which in the most recent reports is the antibody associated in most cases with the development of SP, probably explained by vasculopathic phenomena not only in the skin but also in the lungs, a prevalence of 80%–90% being described in different series, with the remaining 10%–20% being described mainly in anti-synthetase syndrome. These series demonstrate 3 common characteristics in the development of this condition: amyopathic phenotype, ILD, and the presence of anti-MDA5 antibodies, characteristics that were also present in the patients in our cases.8,14–16
There are two important aspects to consider with respect to SP: first, it can present before the classic manifestations of DM or during the course of the disease,3 and second there is a significant impact on prognosis, as mentioned above. However there are different positions regarding the prognostic attribution of this entity, for example, Li et al.17 describe survival rates of 75.4% at one week, 46.2% at 3 months, and 41.9% at one year, without directly attributing the fatal cases to the development of SP, but rather to the concomitance of severe manifestations of inflammatory myopathy per se (pulmonary, muscular, and cutaneous), which also lead to increased use of immunosuppressants, predisposing to an increased risk of severe infections. Highlighting the above idea, Abe et al., in their retrospective cohort, compared patients with DM with and without pneumomediastinum, and found that mortality at one year from initiation of immunosuppressive therapies or optimisation of these therapies was 7.3% in those without pneumomediastinum compared to 34.8% in those with pneumomediastinum; however, more interestingly, individuals with pneumomediastinum were also positive for anti-MDA5 and anti-aminoacyl-tRNA synthetase (65.2% and 26.1%, respectively). In attempting to determine the factors associated with mortality, it was not determined on the presence of one or the other antibody, but on the development of rapidly progressive ILD after performing logistic regression modelling.14 This is in line with our case, where one of the patients died due to rapidly progressive ILD.
The best therapeutic strategy is unclear in the literature and there are no recommendations in the guidelines for the treatment of patients with SP due to DM. The use of different immunosuppressants, such as glucocorticoids, cyclophosphamide, cyclosporine, mycophenolate, immunoglobulin, and even rituximab, has been described; however, the results vary widely, from cases that fully resolved to others with fatal outcomes.5,7,18–21 It is logical to believe, as mentioned above, that the prognosis is more dependent on the development of ILD than on SP per se, requiring only observation until resolution and always bearing in mind that this manifestation may be the harbinger of more severe organ involvement in the short term, as a result of disease activity.8,11,14–16
ConclusionsPatients with anti-MDA5 antibodies are characterised by little or no muscle involvement, but by extensive pulmonary involvement, mainly associated with ILD, which can sometimes be severe enough to result in death.
ILD is not the only pulmonary manifestation in these patients; these 2 cases were noteworthy due to the development of SP, a manifestation that, although atypical, may be a marker of severity associated with worse outcomes. This is why clinicians should be more vigilant and search more thoroughly for the different pulmonary complications described in patients with DM.
Ethical considerationsVerbal and written consent was obtained from the patients, as well as approval from the institution's research and ethics committee.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestsThe authors have no conflict of interests to declare.


