




The clinical and biochemical overlap of various pathologies of phosphocalcic metabolism can lead to misdiagnosis and consequent clinical management. One example is pseudohypoparathyroidism, which can be confused with vitamin D-dependent rickets (VDDR1) if appropriate biochemical determinations are not performed.
Patients and methodsTwo pairs of siblings, from independent families, were clinically diagnosed in adolescence with pseudohypoparathyroidism due to hypocalcaemia, elevated parathyroid hormone levels and normal or elevated phosphorus values. After ruling out alterations in GNAS, a massive sequencing study of genes associated with other differential diagnoses was carried out.
ResultsTwo genetic variants in the CYP27B1 gene potentially associated with the phenotype were identified. Pathogenic variants in this gene are associated with VDDR1A. Clinical-biochemical re-evaluation of the patients confirmed this diagnosis and treatment was adapted.
ConclusionsAlthough VDDR1A is an infrequently diagnosed pathology in adulthood, in cases of hypocalcaemia with elevated PTH values, determination of the 1,25(OH)2D3 and 25(OH)D3 forms of vitamin D is relevant to reach a correct diagnosis.
El solapamiento clínico y bioquímico de diversas enfermedades del metabolismo fosfocálcico puede conllevar un erróneo diagnóstico y su consecuente abordaje clínico. Un ejemplo es el seudohipoparatiroidismo, que puede confundirse con el raquitismo dependiente de vitamina D (VDDR1) si no se hacen las determinaciones bioquímicas adecuadas.
Pacientes y métodosDos parejas de hermanos, de familias independientes, fueron diagnosticados clínicamente en la adolescencia de seudohipoparatiroidismo al presentar hipocalcemia, niveles elevados de hormona paratiroidea y valores normales o elevados de fósforo. Tras descartar alteraciones en GNAS, se realizó un estudio, mediante secuenciación masiva, de genes asociados a otros diagnósticos diferenciales.
ResultadosSe identificaron 2 variantes genéticas en el gen CYP27B1 potencialmente asociadas con el fenotipo. Variantes patogénicas en este gen se asocian con VDDR1A. La reevaluación clínica-bioquímica de los pacientes confirmó dicho diagnóstico y se adecuó el tratamiento.
ConclusionesSi bien la VDDR1A es un trastorno del metabolismo de diagnóstico infrecuente en la edad adulta, en casos de hipocalcemia con valores elevados de PTH es relevante la determinación de las formas 1,25(OH)2D3 y 25(OH)D3 de la vitamina D para alcanzar un diagnóstico correcto.
Pseudohypoparathyroidism (PHP), recently renamed iPPSD2 and 3 (inactivating PTH/PTHrP signalling disorders),1 describes a heterogeneous group of rare genetic or epigenetic diseases characterised by the presence of hypocalcaemia and hyperphosphatemia resulting from the resistance of certain tissues to the biological actions of parathyroid hormone (PTH).2
However, elevated PTH levels are not specific to iPPSD, as other physiological conditions such as hyperparathyroidism, benign parathyroid tumours, renal disease, calcium and phosphorus metabolism disorders or vitamin D deficiency must be ruled out before such a diagnosis.1
We report the cases of 2 pairs of siblings who were diagnosed with pseudohypoparathyroidism in childhood or adolescence. The causal mutation was identified in the CYP27B1 gene after genetic testing in adulthood using next-generation sequencing of a panel of genes associated with iPPSD and other differential diagnoses, suggesting a diagnosis of vitamin D-dependent rickets type 1A.
Patients and methodsFamily 1Case 1A 59-year-old Caucasian woman was referred for genetic counselling due to clinical suspicion of type 2 pseudohypoparathyroidism, diagnosed in childhood, due to the incidental finding of elevated PTH values after an accident with trauma. Currently undergoing treatment with calcium and calcitriol, with elevated PTH associated with vitamin D alterations (Table 1). The patient is short (142 cm;
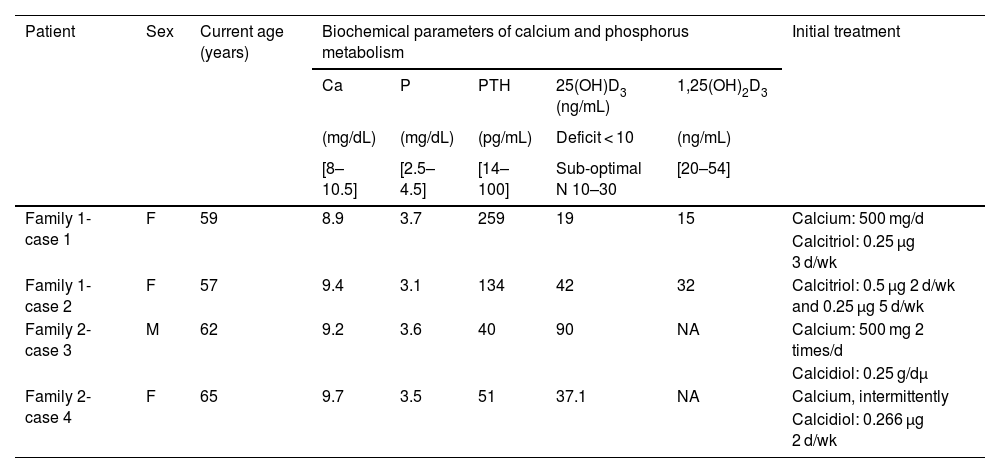
Summary of calcium and phosphorus metabolism data at the time of the patients’ visit to the clinic.
| Patient | Sex | Current age (years) | Biochemical parameters of calcium and phosphorus metabolism | Initial treatment | ||||
|---|---|---|---|---|---|---|---|---|
| Ca | P | PTH | 25(OH)D3 (ng/mL) | 1,25(OH)2D3 | ||||
| (mg/dL) | (mg/dL) | (pg/mL) | Deficit < 10 | (ng/mL) | ||||
| [8–10.5] | [2.5–4.5] | [14–100] | Sub-optimal N 10–30 | [20–54] | ||||
| Family 1-case 1 | F | 59 | 8.9 | 3.7 | 259 | 19 | 15 | Calcium: 500 mg/d |
| Calcitriol: 0.25 μg 3 d/wk | ||||||||
| Family 1-case 2 | F | 57 | 9.4 | 3.1 | 134 | 42 | 32 | Calcitriol: 0.5 μg 2 d/wk and 0.25 μg 5 d/wk |
| Family 2-case 3 | M | 62 | 9.2 | 3.6 | 40 | 90 | NA | Calcium: 500 mg 2 times/d |
| Calcidiol: 0.25 g/dμ | ||||||||
| Family 2-case 4 | F | 65 | 9.7 | 3.5 | 51 | 37.1 | NA | Calcium, intermittently |
| Calcidiol: 0.266 μg 2 d/wk | ||||||||
The treatment they were taking at the time is included.
1,25(OH)2D3: serum levels of 1-alpha,25-dihydroxycholecalciferol or calcitriol; 25(OH)D3: serum levels of 25-hydroxyvitamin D or calcidiol; Ca: serum calcium levels; F: female; mg: milligrams; M: male; NA: not available; P: serum phosphorus levels; PTH: serum parathyroid hormone levels; μg: micrograms, normal range shown in brackets.
A 57-year-old woman, sister of the previous patient, with clinical suspicion of PHP type 2. Despite treatment, she had elevated PTH with normal levels of 1.25(OH)2D3 and 25(OH)D3 (Table 1). Phenotypically, she is at the lower limit of normal height (152 cm; p3–p10) and overweight (61.2 kg; BMI: 26.5). X-rays show a slight shortening of metacarpals II and III (Appendix B Fig. B of the Supplementary material).
Family 2Case 3A 62-year-old man who visited the endocrinology clinic with a clinical diagnosis of pseudohypoparathyroidism at the age of 23 due to hypocalcaemia (5.8 mg/dL; normal range 8−11 mg/dL), hyperphosphatemia (5.4 mg/dL; normal range 2.7–4.5 mg/dL) with normal PTH levels (values not available). Physical examination revealed short stature (150 cm; >p3), normal weight (54.7 kg; BMI: 24.31) and absence of subcutaneous ossifications and brachydactyly.
At the time of medical consultation, the patient was receiving oral treatment with calcidiol and calcium supplements. Laboratory tests revealed normal biochemical parameters (Table 1).
Case 4A 65-year-old woman with clinical suspicion of familial pseudohypoparathyroidism visited an endocrinology clinic at the age of 21 for tetany secondary to hypocalcaemia (5.2 mg/dL), stiffness of hands and circumoral contracture. In addition, bilateral shortening of metacarpals II, III and IV was detected on radiography (Appendix B Fig. C of the Supplementary material) and opacities in the posterior capsule of the lens in both eyes. Throughout the follow-up, elevated PTH levels (101 pg/mL) were occasionally detected.
The existence of the same condition in a sibling (case 3), the biochemical characteristics, and the presence of short stature (136 cm;Table 1).
Molecular studiesEpigenetic and genetic alterations at the GNAS locus were analysed in both families, as already described.3 This was followed by the in-house designed iPPSD-v1 NGS panel testing, in which 92 genes (see Appendix B Supplementary material) associated with iPPSDs and other differential diagnoses were studied. Analysis of the NGS data as well as filtering and prioritisation of variants was carried out as described above.3 Subsequent confirmation and co-segregation studies were performed by Sanger sequencing.
All the studies have been reviewed and approved by the Basque Clinical Research Ethics Committee (CEI-E) and, in addition, the patients have signed the corresponding informed consent for the study to be conducted.
ResultsAfter ruling out epigenetic and genetic alterations at the GNAS locus, the iPPSD-v1 NGS panel testing identified a variant potentially associated with the phenotype in each of the families.
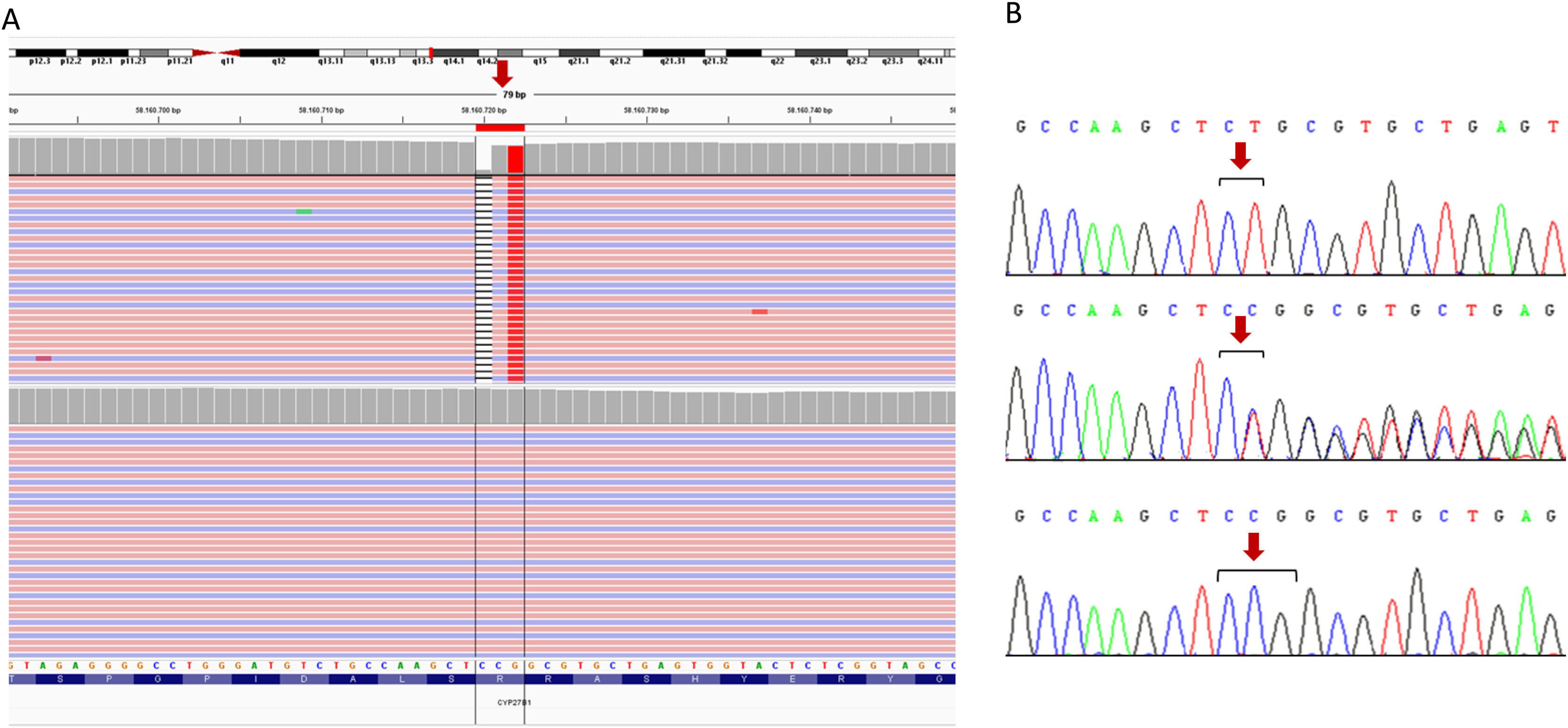
Family 1A probably homozygous variant was identified in exon 1 of the CYP27B1 gene (NM_000785.4: c.103_104delinsA; p.(Ser36Alafs*42)) (Fig. 1). Sanger sequencing in both case 1 and case 2 and in their parents confirmed the presence of the familial variant in homozygosity in both patients and in heterozygosity in their parents. Consanguinity was ruled out.
 Identification of the homozygous mutation in exon 1 of the CYP27B1 gene by next-generation sequencing (NGS). The files obtained from the alignment of the paired-end NGS reads (pink, sense; blue, antisense) are uploaded to the interactive genome viewer, IGV for visualisation. The gene sequence is in the reverse orientation on the chromosome (strand-1). The position of the mutation is indicated by a red arrow. The black stripe represents deletion, subsequently the change is shown by a homozygous thymine (red). The upper panel corresponds to the index person, while the lower panel corresponds to a control person. B) Confirmation by Sanger sequencing of the presence of the c.103_104delinsA variant (named on strand +1, according to international recommendations). The upper panel corresponds to the sequence of the index patient, the middle panel to her father and the lower panel to a control sample. The results for the sister and the mother are identical to those shown in the top and middle panel, respectively (data not shown). Altered nucleotides are shown in black brackets, the position of the mutation is indicated by a red arrow.")
Genetic testing results performed on family 1. A) Identification of the homozygous mutation in exon 1 of the CYP27B1 gene by next-generation sequencing (NGS). The files obtained from the alignment of the paired-end NGS reads (pink, sense; blue, antisense) are uploaded to the interactive genome viewer, IGV for visualisation. The gene sequence is in the reverse orientation on the chromosome (strand-1). The position of the mutation is indicated by a red arrow. The black stripe represents deletion, subsequently the change is shown by a homozygous thymine (red). The upper panel corresponds to the index person, while the lower panel corresponds to a control person. B) Confirmation by Sanger sequencing of the presence of the c.103_104delinsA variant (named on strand +1, according to international recommendations). The upper panel corresponds to the sequence of the index patient, the middle panel to her father and the lower panel to a control sample. The results for the sister and the mother are identical to those shown in the top and middle panel, respectively (data not shown). Altered nucleotides are shown in black brackets, the position of the mutation is indicated by a red arrow.
This is a novel variant, not present in population databases such as gnomAD, ExAC or 1000 Genomes, nor described in the literature associated with the disease. Bioinformatic analysis indicates that the truncated protein is not functional, as the ligand binding sites have been completely eliminated and the vitamin 1α-hydroxylase activity domain has been broken. Therefore, according to ACMG guidelines, this variant is classified as probably pathogenic (PVS1, PM2, PP4).
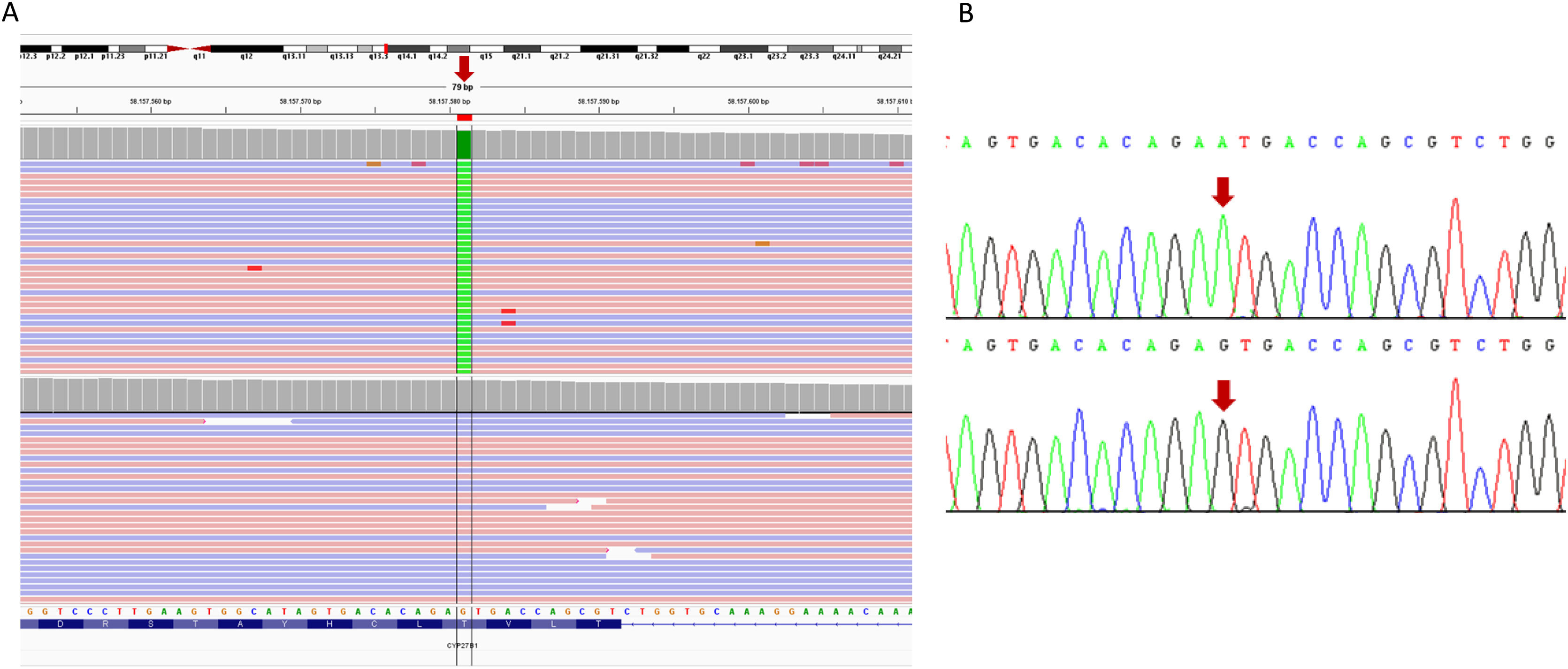
Family 2A probably homozygous variant was identified in exon 8 of the CYP27B1 gene (NM_000785.4: c.1226C>T; p.(Thr409Ile)) (Fig. 2). The algorithm for copy number alteration analysis (VarSeq CNV Caller, Golden Helix, Varseq 2.2.5) showed a ratio of ∼1 (computed based on normalised average coverage versus control samples), which rules out deletions in this gene. The presence of this variant was subsequently confirmed by Sanger sequencing in both patients. No segregation studies were performed as no progenitor sample was available.
 Visualisation in the IGV viewer of the results of the NGS in which the homozygous substitution of guanine for adenine (red arrow) appears in in the CYP27B1 gene in the index patient (upper panel), with respect to a control (lower panel). The gene sequence is in the reverse orientation on the chromosome. B) Validation, by Sanger sequencing, of the presence of the pathogenic variant c.1226C>T (named on strand +1, according to international recommendations). The upper panel corresponds to the sequence of the index patient and the lower panel to a control sample. The arrow indicates the position of the substituted nucleotide.")
Results of the sequencing studies performed on family 2. A) Visualisation in the IGV viewer of the results of the NGS in which the homozygous substitution of guanine for adenine (red arrow) appears in in the CYP27B1 gene in the index patient (upper panel), with respect to a control (lower panel). The gene sequence is in the reverse orientation on the chromosome. B) Validation, by Sanger sequencing, of the presence of the pathogenic variant c.1226C>T (named on strand +1, according to international recommendations). The upper panel corresponds to the sequence of the index patient and the lower panel to a control sample. The arrow indicates the position of the substituted nucleotide.
This variant has already been described as pathogenic in several independent families.4
Clinical reassessmentFamily 1When informed of the genetic result, patient 1 revealed that, in her adolescence, she was bow-legged, had hypocalcaemia and elevated PTH. Currently, she suffers from bone pain, especially in the feet and knees. Bone densitometry shows low bone density in the lumbar spine and proximal femur. The amount and frequency of calcitriol treatment has been modified to 0.5 μg/day to return PTH, 25(OH)D3 and 1,25(OH)2D3 levels to normal and bone pain is currently treated with tapentadol.
Her sister mentions that, in adolescence, she suffered 2 episodes of tetany, had dysplastic teeth and bowed legs. In addition, several blood tests showed hypocalcaemia, hyperphosphatemia, high levels of alkaline phosphatase, PTH and vitamin D deficiency. Tetany was no longer present and vitamin D levels, both 25(OH)D3 and 1,25(OH)2D3, returned to normal after treatment with calcitriol.
Family 2Based on the genetic results, patient 3 was switched from calcidiol to calcitriol (0.25 μg/day), maintaining calcium supplementation. A review of the medical record showed that the patient had suffered seizures at the age of 10, classified as febrile, and at 14 years of age, hypertonia symptoms associated with generalised hypocalcaemia. A bone series study at the age of 23 revealed diffuse osteosclerosis of the spine and femurs with bowing. He currently reports knee osteopenia.
His sister’s treatment was also switched from calcidiol to calcitriol (0.25 μg/day) and she was insisted on the need to take calcium supplements continuously, without success. At 46 years of age, she was suffering from bilateral and intermittent coxofemoral joint pain with no detectable alteration in the bone densitometry study.
DiscussionVitamin D-dependent rickets (VDDR1) is an autosomal recessive disease characterised by deficiency of 1α-hydroxylase activity.5 Pathogenic variants in the CYP27B1 gene are associated with VDDR1A (OMIM #264700), characterised by hypotonia, growth retardation and seizures or tetany. It can also lead to tooth enamel hypoplasia during infancy6 and mild to moderate rickets findings on radiographs. Biochemically, it presents with low serum calcium and phosphorus concentrations together with increased PTH and alkaline phosphatase levels. They associate normal or elevated 25(OH)D3 concentrations with low 1,25(OH)2D3 levels.5
We have identified 2 homozygous variants in the CYP27B1 gene in the present study. Both involve a defect in the enzymatic activity of 1α-hydroxylase, in 2 adult sibling pairs, with intra- and interfamilial clinical variability, which coincide with the doubtful phenotype-genotype correlation in VDDR1A,4 for which it is proposed that factors such as early treatment with variable doses of calcium may mitigate the symptoms.4
Moreover, rPTH is one of the major criteria for the diagnosis of iPPSD.2 However, elevated PTH levels are not always due to resistance, but may be the consequence of a compensatory mechanism triggered by vitamin D deficiency since, when a patient starts to have insufficient or deficient vitamin D levels, there is a compensatory increase in PTH until serum 1,25(OH)2D3 levels return to normal.7 However, in patients with impaired vitamin D metabolism, serum 1,25(OH)2D3 concentrations do not return to normal and PTH values remain elevated.7 For this reason, rPTH is defined as the association of hypocalcaemia, hyperphosphatemia and elevated PTH levels in the absence of vitamin D deficiency.1,2
The patients described in this study had clinical suspicion of PHP/iPPSD because, similar to VDDR1A, they had brachydactyly, hypocalcaemia and elevated PTH levels, and vitamin D concentrations were the differentiating analyte between the two diseases.1,2 Furthermore, in patients with VDDR1A, vitamin D deficiency usually appears after birth and clinical manifestations are usually detected during the first years of life.5 However, severe vitamin D deficiency in adults is less common and usually involves nonspecific clinical manifestations related to osteomalacia, diffuse bone pain and muscle weakness.6 Since there are recommendations that vitamin D levels should not be routinely requested8 and guidelines on optimal vitamin D levels for proper body function have only recently been agreed,9 the differential diagnosis between VDDR1A and iPPSD is complicated.10 However, it has been demonstrated that, in certain cases, the biochemical determination of serum levels of both 1,25(OH)2D3 and 25(OH)D3 can be used to guide and make an appropriate diagnosis.
Ethical considerationsThe studies have been reviewed and approved by the Basque Clinical Research Ethics Committee (CEI-E).
FundingThis study has been funded with partial support from the Instituto de Salud Carlos III of the Ministry of Economy and Competitiveness (Spain), co-financed by the European Regional Development Fund (grant number PI20/00950) and the Department of Health of the Basque Government (grant number GV2021/111056).
Conflict of interestThe authors declare that they have no conflicts of interest.
The following is Supplementary data to this article:
