

Pyruvate kinase deficiency is a hereditary disease that affects the glycolytic pathway of the red blood cell, causing nonspherocytic hemolytic anemia. The disease is transmitted as an autosomal recessive trait and shows a marked variability in clinical expression. This study reports on the molecular characterization of ten Brazilian pyruvate kinase-deficient patients and the genotype–phenotype correlations.
MethodSanger sequencing and in silico analysis were carried out to identify and characterize the genetic mutations. A non-affected group of Brazilian individuals were also screened for the most commonly reported variants (c.1456C>T and c.1529G>A).
ResultsTen different variants were identified in the PKLR gene, of which three are reported here for the first time: p.Leu61Gln, p.Ala137Val and p.Ala428Thr. All the three missense variants involve conserved amino acids, providing a rationale for the observed enzyme deficiency. The allelic frequency of c.1456C>T was 0.1% and the 1529G>A variant was not found.
ConclusionThis is the first comprehensive report on molecular characterization of pyruvate kinase deficiency from South America. The results allowed us to correlate the severity of the clinical phenotype with the identified variants.
Pyruvate kinase (PK) deficiency is the most common enzymatic defect of the glycolytic pathway, causing hereditary nonspherocytic hemolytic anemia. The prevalence of PK deficiency has been estimated to be 1:20000 in the general white population.1 The disease is caused by mutations in the PKLR gene, which are transmitted as an autosomal recessive trait with affected individuals being either homozygotes or compound heterozygotes. The most commonly reported mutations are missense variants, including c.1529G>A in the United States and Northern/Central Europe, c.1456C>T in Southern Europe, and c.1468C>T in Asia.2–7
The severity of anemia is quite variable, ranging from mild or fully compensated forms to life-threatening neonatal anemia that requires continuous transfusions. Other clinical features include jaundice, splenomegaly and gallstones in some patients.8
This study reports on the molecular analysis and the clinical description of PK deficient patients of Brazilian origin.
MethodsPatientsThis study involved ten unrelated patients with PK deficiency originating from Southern Brazil. Patients were either diagnosed at the study center or diagnosed elsewhere and referred to this center for confirmation of the diagnosis and/or to establish the molecular basis of their PK deficiency. Other causes of hemolytic anemia were ruled out in all patients. Appropriate informed consent was obtained either directly, when the patients were over 12 years of age, or from their parents or guardians. The diagnosis was based on clinical history, hematological data, and demonstration of reduced PK activity in red blood cells.
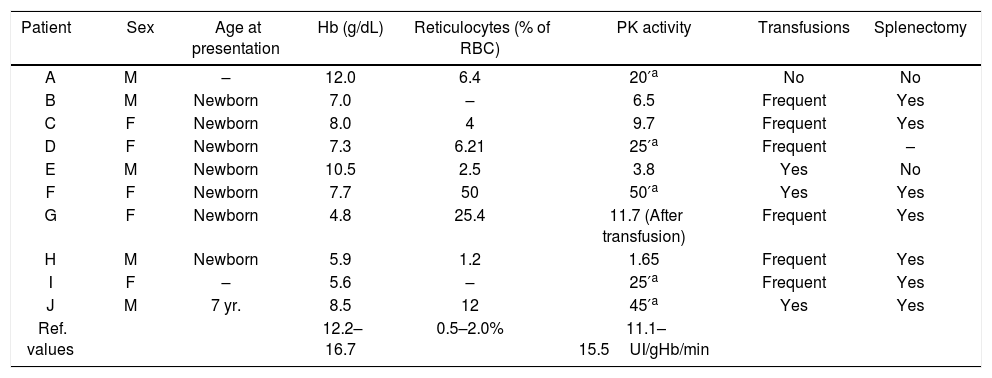
Eight patients (A–H) displayed a severe phenotype hemoglobin levels lower than 8g/dL and/or transfusion dependence and/or splenectomy. Two patients (I and J) displayed a moderate-to-mild phenotype defined by hemoglobin greater than 8g/dL, fewer than five transfusions and no splenectomy (Table 1).
Hematological parameters of Brazilian patients with hemolytic anemia due to PK deficiency.
| Patient | Sex | Age at presentation | Hb (g/dL) | Reticulocytes (% of RBC) | PK activity | Transfusions | Splenectomy |
|---|---|---|---|---|---|---|---|
| A | M | – | 12.0 | 6.4 | 20′a | No | No |
| B | M | Newborn | 7.0 | – | 6.5 | Frequent | Yes |
| C | F | Newborn | 8.0 | 4 | 9.7 | Frequent | Yes |
| D | F | Newborn | 7.3 | 6.21 | 25′a | Frequent | – |
| E | M | Newborn | 10.5 | 2.5 | 3.8 | Yes | No |
| F | F | Newborn | 7.7 | 50 | 50′a | Yes | Yes |
| G | F | Newborn | 4.8 | 25.4 | 11.7 (After transfusion) | Frequent | Yes |
| H | M | Newborn | 5.9 | 1.2 | 1.65 | Frequent | Yes |
| I | F | – | 5.6 | – | 25′a | Frequent | Yes |
| J | M | 7 yr. | 8.5 | 12 | 45′a | Yes | Yes |
| Ref. values | 12.2–16.7 | 0.5–2.0% | 11.1–15.5UI/gHb/min |
Five hundred healthy blood donors from the Blood Center of the Universidade Estadual de Campinas were also studied to analyze the frequency of the c.1456C>T and c.1529G>A variants in a sample Brazilian population.
DNA and structural analysisVenous blood was used for hematological and DNA analysis. Hematological parameters including red blood cell indices were investigated and a reticulocyte count was performed. PK enzyme activity and other biochemical determinations were carried out according to standard methods.9
Total genomic DNA was isolated from peripheral blood leukocytes by the standard salting-out method. Individual exons of the PKLR gene were amplified by polymerase chain reaction (PCR) and DNA sequence analysis was performed using the Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on an ABI 3500xL Genetic Analyzer (Applied Biosystems) with the same primers used in the PCR reactions. The Chromas Lite 2.0 (Technelysium Pty Ltd) and CLC Sequence Viewer v.6.8.1 free software (CLC bio) were used to analyze and compare sequences with the reference PKLR sequence. Structural analyses were performed using the PDB ID: 2VGB – chain A as a template.10 The native and mutant models were constructed by the SWISS MODEL web-served program.11 Internal contacts were evaluated by STING Millennium12 and 3D protein structures were generated using PyMOL (the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
To study the frequency of the c.1529G>A and c.1456C>T variants, exon 10 of the PKLR gene was amplified by PCR. The c.1529G>A variant was screened using the StyI enzyme, which only digests mutated fragments, producing fragments of 144 and 112 base pairs (bp). The c.1456C>T variant was screened using the BsmAI enzyme that only cleaves the DNA of people without these mutations resulting in fragments of 220 and 36bp. The reactions were incubated at 37°C for 16h according to the manufacturer's recommendations and the products were subjected to agarose gel electrophoresis.
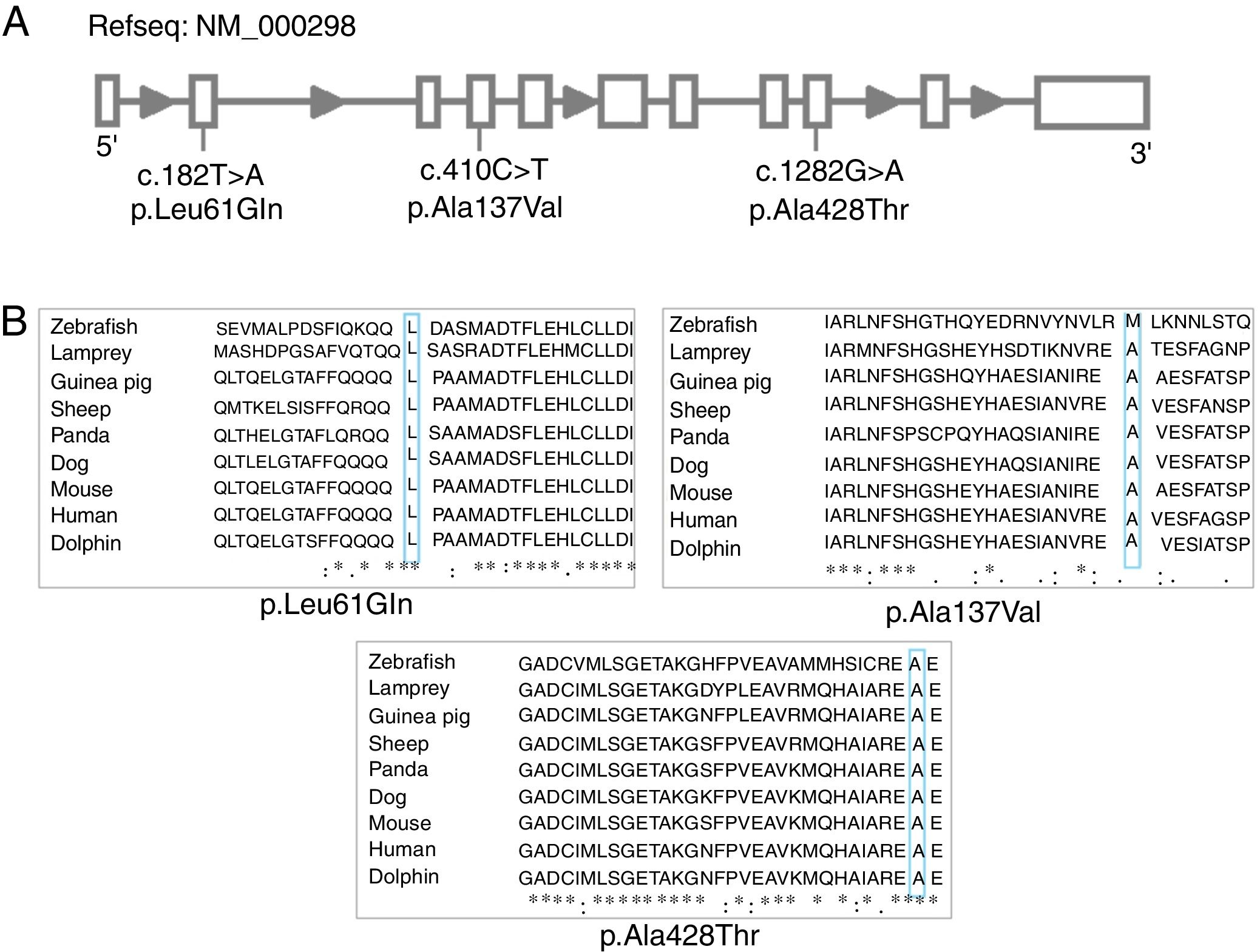
ResultsTen different variants were identified among the ten PK-deficient patients, nine of which are in the coding region (eight missense and one nonsense variants) and one is in the promoter region (Table 2). Seven of these variants had been previously reported: c.721G>T,7 993C>A,13 c.994G>A,14 c.1456C>T,7 c.1511G>T,15 c.1529G>A,7 c.-72A>G.16 The other three are described here for the first time: c.182T>A (p.Leu61Gln), c.1282G>A (p. Ala428Thr) and c.410C>T (p.Ala137Val) (Figure 1A).
Molecular data of 10 PK-deficient patients.
| Patient | Mutation | Effect on protein | Variant classificationb | MAF ExAC (%) |
|---|---|---|---|---|
| A | c.[182T>A]; [1282G>A] | p.[Leu61Gln]; [Ala428Thr] | Uncertain significance; Uncertain significance | Absent; absent |
| B | c.[410C>T]; [410C>T] | p.[Ala137Val]; [Ala137Val] | Uncertain significance | Absent |
| C | c.[1529G>A]; [1529G>A] | p.[Arg510Gln]; [Arg510Gln] | Pathogenic | 0.04 |
| D | c.[1456C>T]; [993C>A] | p.[Arg486Trp]; [Asp331Glu] | Likely pathogenic; uncertain significance | 0.28; 0.03 |
| E | c.[1456C>T]; [−72A>G] | p.[Arg486Trp]; Transcriptional mutation | Likely pathogenic; likely pathogenic | 0.28; not provided |
| F | c.[1511G>T]; [−72A>G] | p.[Arg504Leu]; Transcriptional mutation | Pathogenic; likely pathogenic | 0.0008/not provided |
| G | c.[994G>A]; [994G>A] | p.[Gly332Ser]; [Gly332Ser] | Pathogenic | 0.006 |
| H | c.[721G>T]; [721G>T] | p.[Glu241a]; [Glu241a] | Pathogenic | 0.003 |
| I | c.[1511G>T]; [1511G>T] | p.[Arg504Leu]; [Arg504Leu] | Pathogenic | 0.0008 |
| J | c.[1456C>T]; [?] | p.[Arg486Trp]; [?] | Likely pathogenic | 0.28 |
 Linear representation of the gene with variants located. (B) Multiple alignment of native PK-R protein using ClustalW. The residues L61, A428 and A137 are highlighted. Asterisks indicate amino acid residues conserved throughout the nine species.")
All the new missense variants described appear to be damaging/disease causing according to computational predictions: Polyphen-2,17 Sift18 and Mutation Taster.19 The three new variants were not detected in the DNA of the 80 normal controls studied.
The c.182T>A (p.Leu61Gln) and c.1282G>A (p.Ala428Thr) variants were identified in a heterozygous state in Patient A, a 21-year-old male. He had a mild phenotype, without the need of transfusions or splenectomy (Table 1). The patient presented with altered blood cell counts and splenomegaly. He has macro and microcytic hemolytic anemia, with the presence of target cells and poikilocytosis [Hemoglobin (Hb): 12.0g/dL; mean corpuscular volume: 91.7 fl; mean corpuscular hemoglobin: 30.3 pg; mean corpuscular hemoglobin concentration: 33.1g/dL; red blood cell distribution width: 13.6%; reticulocytes: 6.4%; ferritin: 525.1ng/mL; haptoglobin less than 7.9ng/dL and unconjugated hyperbilirubinemia]. The patient also presented with the sickle cell trait (Hb S: 40%).
The c.410C>T (p.Ala137Val) variant was identified in homozygosis in Patient B, a 14-year-old male. The patient presented with a severe phenotype requiring transfusions since the first month of life. His parents are first-degree cousins; they lost their first child shortly after birth due to severe anemia.
Structural analyses of the new variants were obtained. The wild-type residue of the three variants is conserved among several species (Figure 1B). Neither the mutant residues nor another residue type with similar properties was observed in these positions in other homologous sequences. The three new variants are in regions reported in UniProt to form α-helixes. The variants did not induce significant conformational changes in the overall protein conformation and, therefore, their description of the mutant structures is restricted here mainly to the sites affected by the variants.
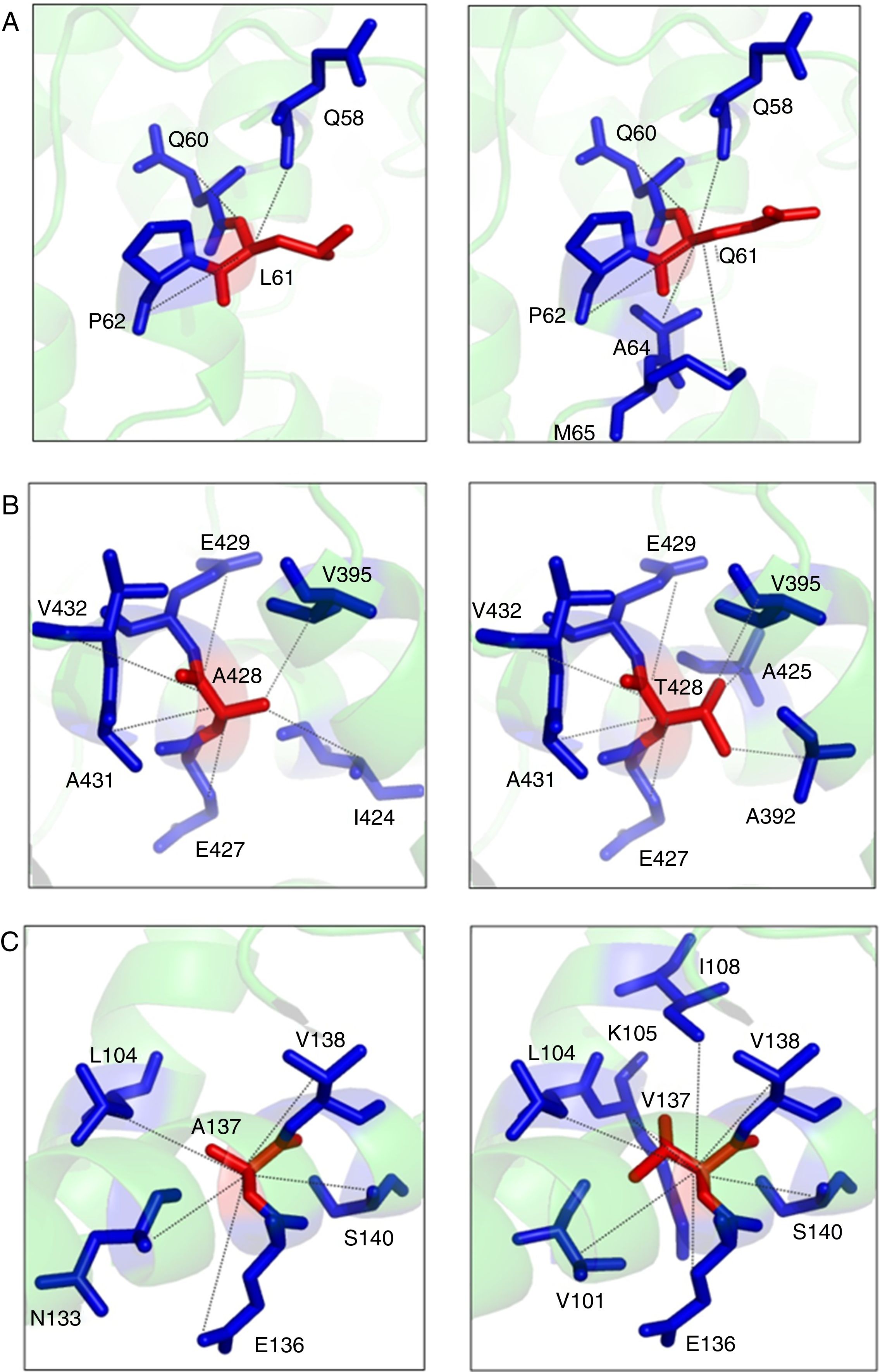
The p.Leu61Arg variant results in a change in the protein conformation at the N domain interface. The residue is located on the surface of the protein. The hydrophobicity of the wild type and mutant residue differ and could cause loss of hydrophobic interactions with other molecules. The 3D representation of the normal and mutant amino acids shows that the mutant residue creates contacts with two additional amino acids of the protein (Figure 2A).
 Structural model of the native and mutant (p.Leu61Arg) PK-R protein showing internal contacts. Native protein (left): the amino acid L61 interacts by hydrogen bond with Q58 and by hydrophobic interaction and hydrogen bond with Q60 and P62. Mutant protein (right): the Q61 establishes new hydrophobic contacts with A64 and M65. (B) Structural model of the native and mutant (p.Ala428Thr) PK-R protein showing internal contacts. Native protein (left): the amino acid A428 interacts by hydrogen bond with V432, A431 and I424, by hydrophobic interaction and hydrogen bond with E429 and E427 and by hydrophobic interaction with V395. Mutant protein (right): the T428 establishes new hydrogen bond contact with A425 and hydrophobic interaction contact with A392; the interaction with E429 changes for hydrophobic interaction and hydrogen bond, and the interaction with I424 is lost. (C) Structural model of the native and mutant (p.Ala137Val) PK-R protein showing internal contacts. Native protein (left): the amino acid A137 interacts by hydrogen bond with S140 and N133, by hydrophobic interaction and hydrogen bond with V138 and E136 and hydrophobic interaction with L104. Mutant protein (right). The V137 establishes new hydrophobic contact with I108, K105 and V101, changes the interaction in S140 for hydrogen bond and loses the interaction with N133.")
(A) Structural model of the native and mutant (p.Leu61Arg) PK-R protein showing internal contacts. Native protein (left): the amino acid L61 interacts by hydrogen bond with Q58 and by hydrophobic interaction and hydrogen bond with Q60 and P62. Mutant protein (right): the Q61 establishes new hydrophobic contacts with A64 and M65. (B) Structural model of the native and mutant (p.Ala428Thr) PK-R protein showing internal contacts. Native protein (left): the amino acid A428 interacts by hydrogen bond with V432, A431 and I424, by hydrophobic interaction and hydrogen bond with E429 and E427 and by hydrophobic interaction with V395. Mutant protein (right): the T428 establishes new hydrogen bond contact with A425 and hydrophobic interaction contact with A392; the interaction with E429 changes for hydrophobic interaction and hydrogen bond, and the interaction with I424 is lost. (C) Structural model of the native and mutant (p.Ala137Val) PK-R protein showing internal contacts. Native protein (left): the amino acid A137 interacts by hydrogen bond with S140 and N133, by hydrophobic interaction and hydrogen bond with V138 and E136 and hydrophobic interaction with L104. Mutant protein (right). The V137 establishes new hydrophobic contact with I108, K105 and V101, changes the interaction in S140 for hydrogen bond and loses the interaction with N133.
The p.Ala428Thr variant results in a change in the protein conformation at the A domain interface, the most highly conserved region. The mutant residue probably lost contact with one amino acid and gained contact with two other residues of the protein (Figure 2B).
The p.Ala137Val wild type probably lost contact with one amino acid and gained contact with three other residues of the protein (Figure 2C).
DiscussionThe clinical picture of the patients was heterogeneous, ranging from mild chronic hemolytic anemia to severe anemia presenting at birth and requiring multiple transfusions. Analysis of the correlation between molecular defect and disease severity in large series of patients has shown that the heterogeneity depends mainly on the type and position of the variant affecting the PKLR gene.20,7,21,13
Studies in PK-deficient patients found that c.1529G>A was the most common variant in Central/Northern Europe and in the United States.6,13,14,22 However, in Southern/Western European patients the most common variant appears to be c.1456C>T.21,5,2 In this sample of PK deficient patients, the c.1456C>T and c.1511G>T variants had similar frequencies (15%).
The frequency of c.1456C>T in this sample is in accordance with that expected in Latino populations. In 500 normal individuals of southeastern Brazil screened in this study, the c.1456C>T variant was identified in 0.1% of the alleles and the c.1529G>A variant was not found. According to the Exome Aggregation Consortium (http://exac.broadinstitute.org/), the frequency of these alleles in Latino populations are 0.17% and 0%, respectively. In the general population, the frequencies are 0.28% and 0.04%, respectively.
The new variants (c.182T>A and c.1282G>A) were identified in heterozygous in Patient A, who displayed a mild clinical phenotype. The c.182T>A (p.Leu61Gln) variant is in the N domain of the protein and could impair hydrophobic interactions with other molecules. Our prediction shows that the mutant residue creates additional contacts with other amino acids, and may perturb the structure of the protein. The c.1282G>A (p. Ala428Thr) variant is in the A domain and is likely to perturb the hydrophobic interior of the protein, forcing the surrounding side chains to rearrange thereby affecting protein stability and/or function.
The c.410C>T (p.Ala137Val) variant, located at the A domain interface of the protein, was identified in homozygosis. Several studies have demonstrated that variants in the A domain, especially in the homozygous form, are associated with severe hemolytic anemia,2,10,21,23 such as in the case of Patient B in this study. The mutant residue is bigger and probably lost contact with one amino acid and gained contacts with additional residues.
In the present study, the c.1529G>A variant was identified in the homozygous state in Patient C, a female child, who presented with a severe clinical phenotype. This variant was previously reported in homozygosis and was also associated with a severe clinical presentation.3
Analysis of the PKRL gene in Patient D showed the presence of biallelic variants (c.993C>A and c.1456C>T). This patient presented with a severe clinical phenotype requiring frequent transfusions and a hemoglobin level less than 8g/dL. These two variants were also identified previously in compound heterozygosity in an Italian patient with a mild phenotype.24 The variability of clinical expression may be explained by individual differences in metabolic or proteolytic activity, or even the ability to compensate for the enzyme deficiency by overexpressing isozymes or using alternative pathways.
This study also reports two patients heterozygous for an A>G substitution at nucleotide −72 relative to the initiation codon of the PKLR gene.25 It was shown that this variant affects the normal promoter transcriptional activity of the PKLR gene resulting in a severe reduction at the PKLR mRNA level. The two patients with the variant reported in this study presented with distinct phenotypes. Analysis of the PKRL gene in Patient E showed the presence of a compound heterozygosity state for the c.-72 A>G and c.1456 C>T variants; the patient presented with a mild phenotype. This result is consistent with previous findings that also describe a mild phenotype in the presence of these two changes in heterozygosis.16 Patient F, on the other hand, presented with the c.-72 A>G in compound heterozygosity with the c.1511G>T variant and expresses a severe phenotype.
Patient G presented with a severe phenotype including an extremely low hemoglobin level and frequent transfusions. The molecular analysis revealed homozygosity for the c.994G>A variant. Previous studies also show that this variant in homozygosis leads to a severe anemia with the need of regular transfusions.21,14
The nonsense variant, c.721G>T, was found in Patient H in homozygosis. This variant causes the truncation of the protein in the Glu241 amino acid and thus explains the severity of the phenotype.
The c.1511G>T variant causes extreme instability of the protein26 with severe anemia in PK-deficient patients homozygous for this variant.15 This variant was found in Patient I, who presented with a severe clinical phenotype.
Patient J carried only one variant in heterozygosis, giving a genotype that could not explain the phenotype. One hypothesis is that the anemia was the result of an association with another red blood cell disease. Further studies are required to clarify the effect of such associations on the phenotype.
This is the first comprehensive report on the molecular characterization of PK deficiency in South America. Ten cases were characterized and three new variants were identified. The etiology was elucidated in nine patients. Collectively, the results contribute to a better understanding of the genotype-to-phenotype correlation in PK deficiency.
Conflicts of interestThe authors declare no conflicts of interest
The authors are grateful to all the members of the staff of our laboratory, and the patients and families participating in this study for their close cooperation and important contributions.


