
To determine the mortality rate of children, adolescents and adults with sickle cell anemia in Rio de Janeiro, Brazil.
MethodsThe number of deaths, the mortality rate and the causes of deaths in patients with sickle cell anemia who were treated and followed up at our institution for 15 years were determined and compared to data available for the Brazilian population.
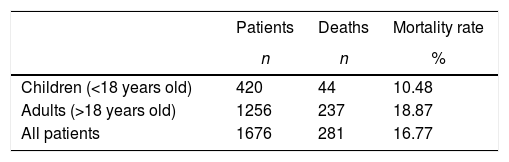
ResultsThe overall number of deaths was 281 patients with a mortality rate of 16.77%. Survival probability was significantly higher in females. The number of deaths and the mortality rate were age-specific with a significant increase in the 19- to 29-year-old age group. The remaining life expectancy of the patients with sickle cell anemia was less than that of Brazilians at large. The gap between the two was about 20 years for ages between one and five years with this gap decreasing to ten years after the age of 65 years. The most common causes of death were infection, acute chest syndrome, overt stroke, organ damage and sudden death during painful crises.
ConclusionTo the best of our knowledge, this is the first Brazilian study in a single institution in Rio de Janeiro; the mortality rate was 18.87% among adult patients with sickle cell anemia. The mortality rates in children and adults are higher than those reported in developed countries of the northern hemisphere.
The mortality rate among infants, children and young adults with sickle cell disease (SCD) in general and sickle cell anemia (SCA) in particular is high. The overall death rates in under 20-year-old children reported by the Cooperative Study of Sickle Cell Disease (CSSCD) about 30 years ago were 2.6% for patients with all types of SCD and 3.3% for patients with SCA.1 Sickle cell disease-related mortality decreased significantly from 1999 to 2009 in the USA in all pediatric age groups up to age 19 years.2 This decline in mortality was the highest for children younger than one year where the decrease was 61% less than the mortality rate during 1979–1998.2 The mortality in young adults (ages 20–24 years), however, increased sharply compared to the mortality in children ages 15–19 years from 0.6/100,000 to 1.4/100,000.2 This increase is attributed to problems associated with the transition from pediatric to adult care.
Unlike the USA, the mortality rate in developing countries continues to be high especially in Africa.3,4 In the state of Minas Gerais, Brazil the mortality rate among children with SCD born during 2009–2011 was not significantly different from that for children born during 1999–2001 (6.2% vs. 5.8%).5 This suggests that newborn screening alone is not enough to reduce the mortality rate of children with SCA. Regular follow-ups, compliance to treatment, family support and the style of life are important factors to maintain the decreases in both morbidity and mortality rates. The purpose of this study was to determine the mortality rates and the cause of death in children, adolescents and adults with SCA followed-up at a single institution in Rio de Janeiro and compare these rates to Brazilian children and adults at large.
MethodsPatientsData of patients with SCA including patients with sickle/β0-thalassemia (S/β0thal), who were seen and followed at HEMORIO for 15 years from January 1, 1998 through December 31, 2012 were retrospectively collected and analyzed. Children (<18 years old) and adults (≥18 years) were included in the study. Age and sex related mortality were also determined and compared to available data of the Brazilian population at large.
The diagnosis of SCA including S/β0thal was confirmed by high performance liquid chromatography (HPLC) as previously described.6 The date and cause of deaths were confirmed on patients’ charts if death occurred at HEMORIO. Death outside HEMORIO, suspected when patients failed to show up for a follow-up, was confirmed by interviews with the patient's families and from death certificates. Accordingly, the death of 69 patients occurred outside HEMORIO. The family brought the death certificates of 62 patients. Of these 13 deaths were due to accidents unrelated to SCA and in 49 patients, death was due to stroke, acute chest syndrome (ACS) or infection. The cause of death in the remaining seven patients was not clear or could not be confirmed and hence, their cause of death was included in the unknown category.
The study was approved by the Institutional Review Board of HEMORIO.
Statistical analysisStatistical analysis was performed using the R software7 and the Kaplan–Meier estimator to assess the overall survival of patients with SCA. Comparison of survival curves was made using the Tarone-Ware test.8 Exclusions from statistical analyses included patients whose deaths were unrelated to SCD and those whose cause of death was unknown. Comparisons of frequencies between nominal variables were made using the chi-squared test without continuity correction or the Fisher exact test. Determination of the average remaining life expectancy was determined by the empirical likelihood (Emplik) ratio test package9 of the R software. Statistical significance was based on a p-value <0.05.
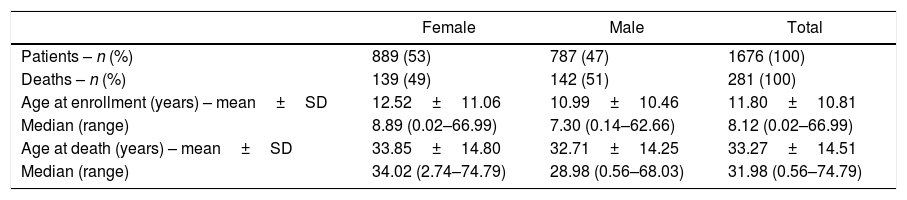
ResultsThe total number of patients enrolled in this study was 1676. Age (both at enrollment and at death) and sex are summarized in Table 1. A total of 281 patients died during the study period (Table 1). The mortality rate is summarized in Table 2.
Characteristics of patients with sickle cell anemia.
| Female | Male | Total | |
|---|---|---|---|
| Patients – n (%) | 889 (53) | 787 (47) | 1676 (100) |
| Deaths – n (%) | 139 (49) | 142 (51) | 281 (100) |
| Age at enrollment (years) – mean±SD | 12.52±11.06 | 10.99±10.46 | 11.80±10.81 |
| Median (range) | 8.89 (0.02–66.99) | 7.30 (0.14–62.66) | 8.12 (0.02–66.99) |
| Age at death (years) – mean±SD | 33.85±14.80 | 32.71±14.25 | 33.27±14.51 |
| Median (range) | 34.02 (2.74–74.79) | 28.98 (0.56–68.03) | 31.98 (0.56–74.79) |
SD: standard deviation.
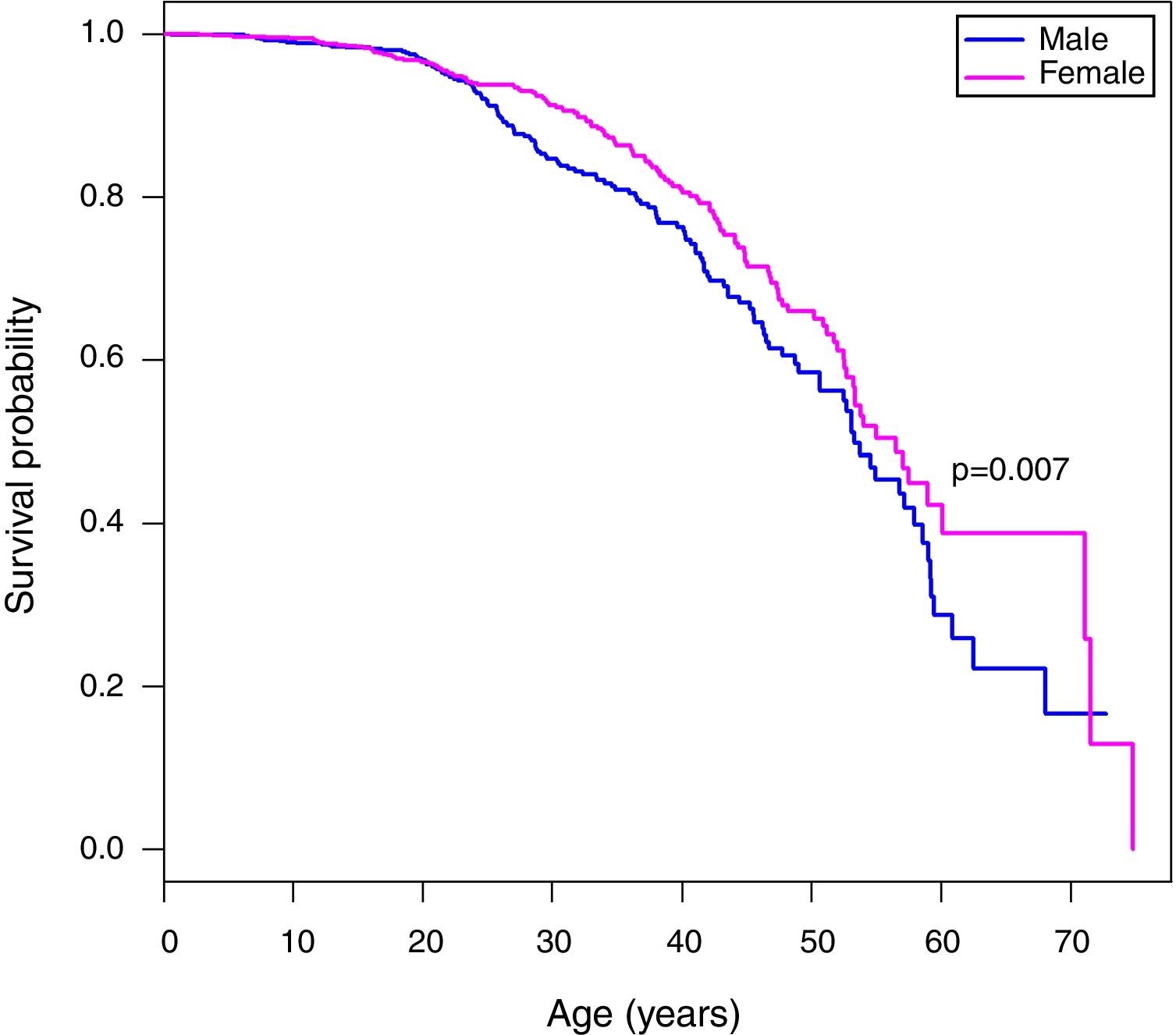
A Kaplan Meier analysis8 of the survival probability of all enrolled patients showed that survival of females was significantly higher than males (p-value=0.007). The median age of survival was 53.3 years for males and 56.5 years for females (Figure 1). The median age at death was 28.98 years for men and 34.02 years for women (Table 1).
.")
Kaplan–Meier curves of patients with sickle cell anemia stratified by sex. The median age of survival of males is 53.3 years, while for females the median survival is 56.5 years. The Tarone-Ware test indicates a statistically significant difference between the curves (p-value=0.007).
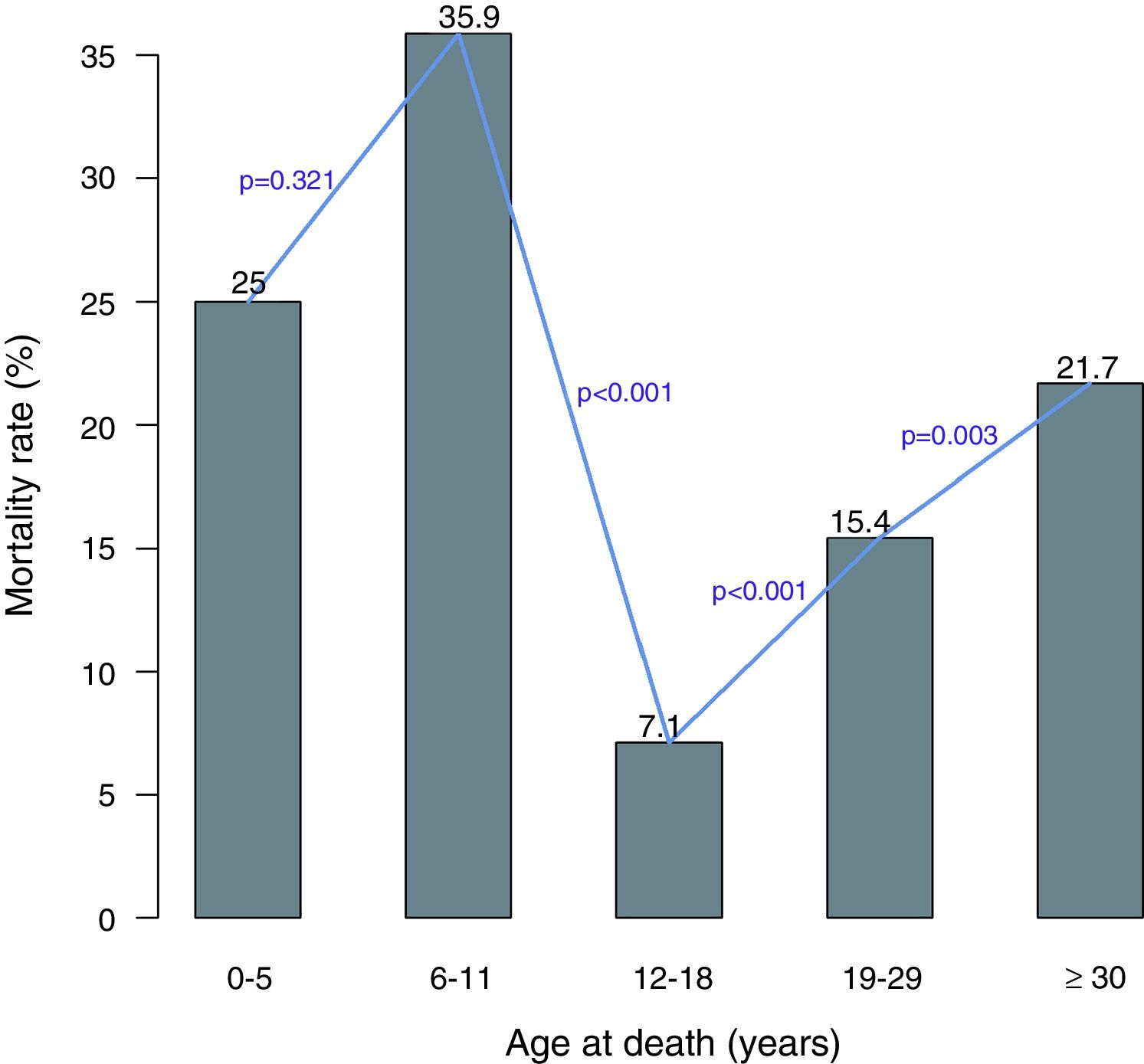
The mortality among patients varied significantly with age. As shown in Figure 2 the mortality rate in under 5-year-old patients was lower than the mortality rate in the 6–11 age group. However, there was a significant difference in the mortality rates between the 6–11 and 12–18 age groups. After the age of 18 years, there was a significant and sudden increase in the mortality in the 19–29 age group. The mortality continued to increase significantly after the age of 30 years.

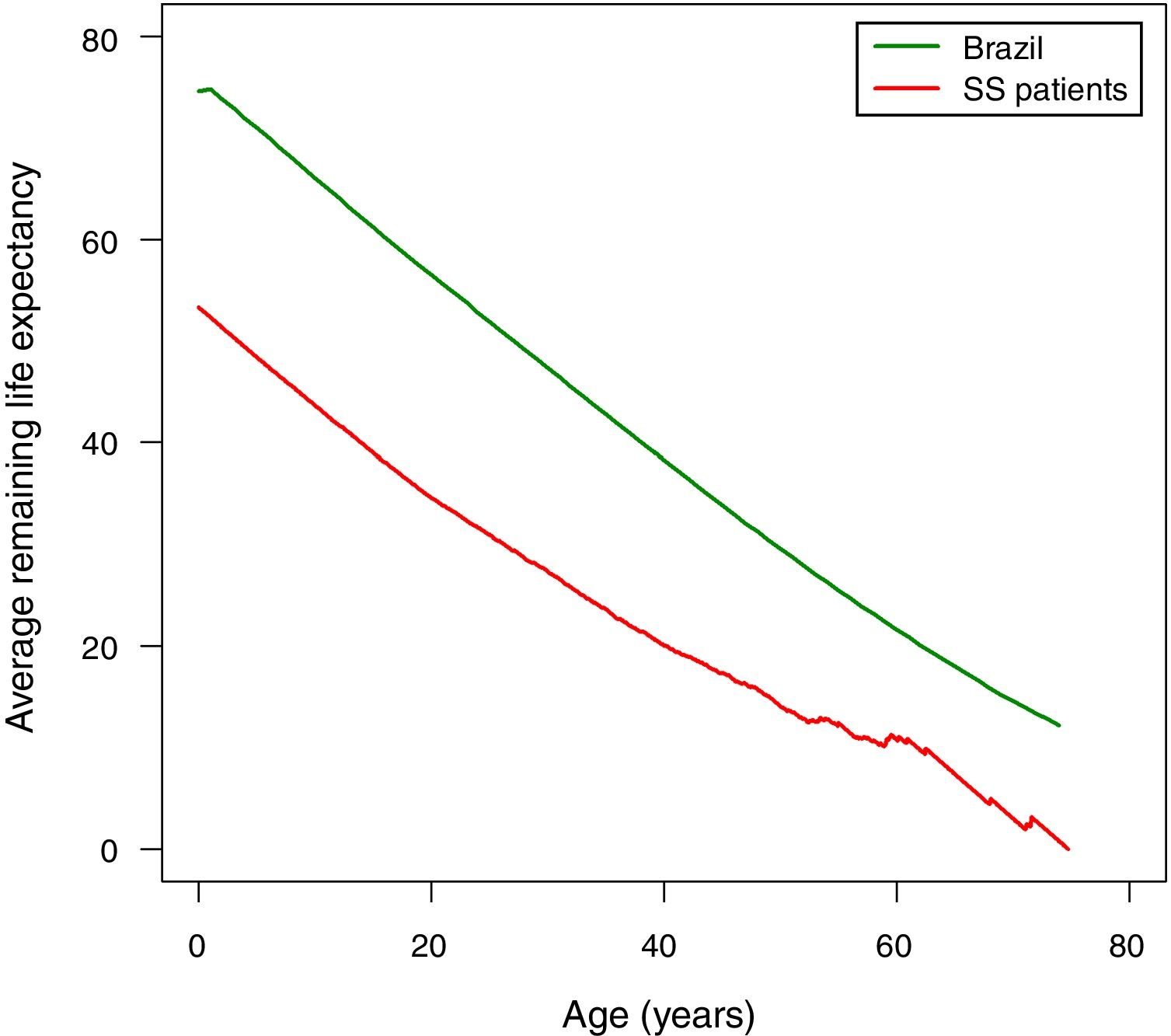
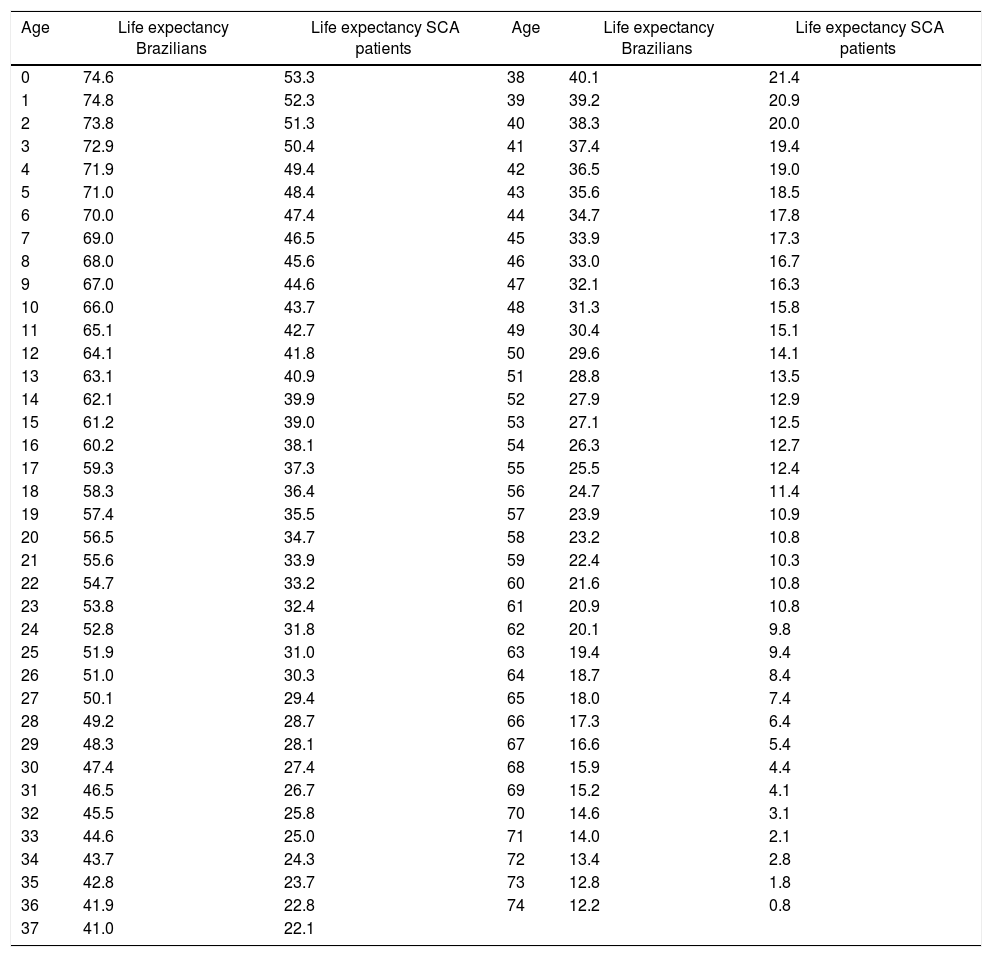
The following assumptions were made in order to compare the life expectancy of patients with SCA in Rio de Janeiro with the life expectancy of Brazilians at large. The prevalence of SCD in the state of Rio de Janeiro is the second highest in Brazil after the state of Minas Gerais. In 2016, about 4000 patients with SCD were treated at HEMORIO representing about 80% of the patients with SCD in the state of Rio de Janeiro. Thus, by extrapolation we assumed that the life expectancy of patients with SCA in Rio de Janeiro may represent the life expectancy of patients with SCA in Brazil. We next compared the life expectancy of patients with SCA described in this study with the life expectancy in Brazilians at large (Figure 3). The latter was based on the 2013 life expectancy report of Brazilians published by the Instituto Brasileiro de Geografia e Estatistica (IBGE).10 It is important to note that life tables are used to measure the mortality, survival, and remaining lifetime of a population at various ages as previously described.11,12

Accordingly, the life expectancy of Brazilian males is 71.0 years and for females it is 78.3 years and the estimated life expectancy for both sexes at birth is 74.6 years (Table 3). The remaining life expectancy at birth of the patients with SCA is 53.3 years, that is, 21.3 years less than the life expectancy of Brazilians at large. Between the ages of one and five years, the remaining life expectancy of the patients in this study is about 22 years less than that of Brazilians at large. This gap decreased to about ten years after the age of 60 as shown in Table 3.
Average remaining life expectancy of sickle cell anemia (SCA) patients compared to that of Brazilians at large.
| Age | Life expectancy Brazilians | Life expectancy SCA patients | Age | Life expectancy Brazilians | Life expectancy SCA patients |
|---|---|---|---|---|---|
| 0 | 74.6 | 53.3 | 38 | 40.1 | 21.4 |
| 1 | 74.8 | 52.3 | 39 | 39.2 | 20.9 |
| 2 | 73.8 | 51.3 | 40 | 38.3 | 20.0 |
| 3 | 72.9 | 50.4 | 41 | 37.4 | 19.4 |
| 4 | 71.9 | 49.4 | 42 | 36.5 | 19.0 |
| 5 | 71.0 | 48.4 | 43 | 35.6 | 18.5 |
| 6 | 70.0 | 47.4 | 44 | 34.7 | 17.8 |
| 7 | 69.0 | 46.5 | 45 | 33.9 | 17.3 |
| 8 | 68.0 | 45.6 | 46 | 33.0 | 16.7 |
| 9 | 67.0 | 44.6 | 47 | 32.1 | 16.3 |
| 10 | 66.0 | 43.7 | 48 | 31.3 | 15.8 |
| 11 | 65.1 | 42.7 | 49 | 30.4 | 15.1 |
| 12 | 64.1 | 41.8 | 50 | 29.6 | 14.1 |
| 13 | 63.1 | 40.9 | 51 | 28.8 | 13.5 |
| 14 | 62.1 | 39.9 | 52 | 27.9 | 12.9 |
| 15 | 61.2 | 39.0 | 53 | 27.1 | 12.5 |
| 16 | 60.2 | 38.1 | 54 | 26.3 | 12.7 |
| 17 | 59.3 | 37.3 | 55 | 25.5 | 12.4 |
| 18 | 58.3 | 36.4 | 56 | 24.7 | 11.4 |
| 19 | 57.4 | 35.5 | 57 | 23.9 | 10.9 |
| 20 | 56.5 | 34.7 | 58 | 23.2 | 10.8 |
| 21 | 55.6 | 33.9 | 59 | 22.4 | 10.3 |
| 22 | 54.7 | 33.2 | 60 | 21.6 | 10.8 |
| 23 | 53.8 | 32.4 | 61 | 20.9 | 10.8 |
| 24 | 52.8 | 31.8 | 62 | 20.1 | 9.8 |
| 25 | 51.9 | 31.0 | 63 | 19.4 | 9.4 |
| 26 | 51.0 | 30.3 | 64 | 18.7 | 8.4 |
| 27 | 50.1 | 29.4 | 65 | 18.0 | 7.4 |
| 28 | 49.2 | 28.7 | 66 | 17.3 | 6.4 |
| 29 | 48.3 | 28.1 | 67 | 16.6 | 5.4 |
| 30 | 47.4 | 27.4 | 68 | 15.9 | 4.4 |
| 31 | 46.5 | 26.7 | 69 | 15.2 | 4.1 |
| 32 | 45.5 | 25.8 | 70 | 14.6 | 3.1 |
| 33 | 44.6 | 25.0 | 71 | 14.0 | 2.1 |
| 34 | 43.7 | 24.3 | 72 | 13.4 | 2.8 |
| 35 | 42.8 | 23.7 | 73 | 12.8 | 1.8 |
| 36 | 41.9 | 22.8 | 74 | 12.2 | 0.8 |
| 37 | 41.0 | 22.1 |
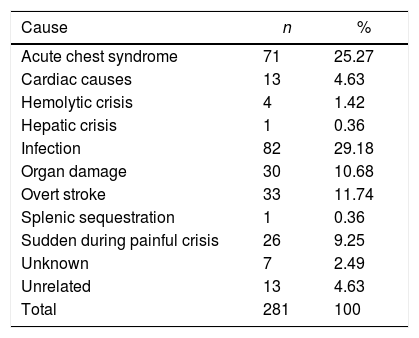
The causes of death of the 281 patients who died are listed in Table 4. The most common causes of death were infection, mostly due to sepsis, ACS, overt stroke, sudden death during painful crises and organ damage due to hepatic or renal failure. Thirteen patients died of unrelated causes mostly due to trauma.
Causes of death of the patients with sickle cell anemia.
| Cause | n | % |
|---|---|---|
| Acute chest syndrome | 71 | 25.27 |
| Cardiac causes | 13 | 4.63 |
| Hemolytic crisis | 4 | 1.42 |
| Hepatic crisis | 1 | 0.36 |
| Infection | 82 | 29.18 |
| Organ damage | 30 | 10.68 |
| Overt stroke | 33 | 11.74 |
| Splenic sequestration | 1 | 0.36 |
| Sudden during painful crisis | 26 | 9.25 |
| Unknown | 7 | 2.49 |
| Unrelated | 13 | 4.63 |
| Total | 281 | 100 |
Although previous studies determined the mortality rate in Brazilian children with SCA, to the best of our knowledge this is the first study that determined the mortality rate in Rio de Janeiro and probably, by extrapolation, in Brazilian adults with SCA. The overall 10.48% mortality rate of all children in this study is higher than that reported in the USA and Europe1,2,13 but similar to other reports on Brazilian children.5,14 Moreover, the Minas Gerais study reported no significant difference in the mortality rate in 1999–2001 after the initiation of their newborn screening program in 1998 compared to that in 2009–2011.5
On comparing age groups, after the significant decrease in the mortality rate in children aged 6–11 years and in adolescents, there was a sudden and sharp increase in the mortality rate in patients aged 19–29 years similar to that reported in the USA. This surge in mortality after adolescence is not well understood and usually it is assumed to be due to a number of issues related to the psychosocial domain.2,15 However, this sudden increase in mortality in patients with SCD aged >18 years of age was not present in a Belgian study that evaluated the survival of patients with SCD recorded in the SCD registry.13
As reported in previous studies, the mortality rate in children is similar between males and females.1,2,16,17 The divergence in the mortality rate between the sexes in favor of females becomes apparent in adults as shown in this report as well as in previous studies.1,18 The reason why adult women with SCA live longer than males is not known. One possibility is that Hb F may be higher in females due to the partial control of Hb F by an X-linked gene located at Xp22.2.19–21 Another possibility is the relatively lower blood viscosity due to lower Hb and hematocrit levels in women.
In order to determine the significance of the mortality rate of patients with SCA, the data of this study were compared with what is known about mortality in children and the life expectancy in the Brazilian population. The last mortality rate for all children <5 years of age in Brazil reported by the United Nations Children's Fund (UNICEF)22 was 1.6% for males and 1.3% for females. These rates are 14.3% for males and 33.3% for females with SCA in the current study. Thus, the mortality rates for under 5-year-old patients with SCA in Rio de Janeiro are about 8.9 times and 25.6 times higher for males and females, respectively. Moreover, the mean difference in average remaining life expectancy for a Brazilian patient with SCA in Rio de Janeiro is 17.6 years (range: 10.0–22.6 years) less than that in the general Brazilian population.7,9,10 The average life expectancy for Brazilians at birth is 74.6 years. The mean survival of Brazilian patients with SCA is 53.3 years, 21.3 years less than the general population.
Among the known causes of death due to SCA, infection was the most common cause of death in both children and adults followed by ACS. The most common causes of death in the Brazilian population at large are heart problems, stroke, pneumonia, chronic obstructive pulmonary disease and diabetes mellitus.23
In conclusion, to the best of our knowledge this is the first Brazilian study that reports the mortality rate among adult patients with SCA in Rio de Janeiro. Unfortunately, this mortality rate is higher than that reported in developed countries in the northern hemisphere. Similarly, the mortality rate among Brazilian children with SCA is higher than that in developed countries. We hope that the mortality rate in Brazilian children and adults will decrease with time as the experience of providers and logistic issues of the newborn screening program improve. Currently, newborn screening seems to be necessary but not sufficient to reduce the mortality rate of children with SCA. In addition, other factors such as regular follow-ups, compliance to treatment, family support and the style of life are important factors to decrease both morbidity and mortality.
Conflicts of interestThe authors declare no conflicts of interest.
Supported in part by the Office of the Secretary of Health for the State of Rio de Janeiro and the Brazilian Ministry of Health.


