
Hereditary hemoglobinopathies, the most common monogenic hemoglobin (Hb) disorders, result in a variety of clinical consequences. It has been observed that various Hb variants and thalassemias are found common to specific ethnic groups and regions. Hb DIran is a structural Hb variant resulting from the substitution of glutamine with glutamate at codon 22 (GAA>CAA, Glu>Gln) of the beta globin gene. This Hb variant was first reported by Rahbar in 1973 in a family from the central part of Iran.1 A deletion of four bases in codon 41/42 (-CTTT) is a rare β0-thalassemia mutation reported in India with a prevalence of 3–15%.2 The present report describes a rare combination of these two mutations for the first time in India.
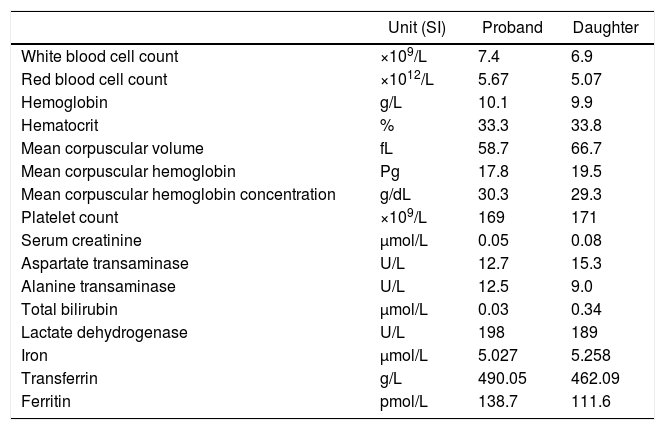
Case reportA 45-year-old Sikh female from Sundergarh district of Odisha, India with a family history of β-thalassemia attended the Sickle Cell Institute, VIMSAR, Burla to screen her status. She was asymptomatic and had no history of blood transfusion or vaso-occlusive crisis. Ultrasonographic examination revealed normal spleen and liver. The various investigations of the proband and her daughter, including a complete blood count and biochemistry, are shown in Table 1. As evident, the index case had features suggestive of microcytic hypochromic anemia (mean corpuscular volume: 58.7fL and mean corpuscular hemoglobin: 17.8pg). An iron profile study indicated possible iron overload [iron 5.027mg/dL (reference range – RR: 0.005–0.175mg/dL); ferritin: 138.7μg/L (RR: 20–200μg/L) and transferrin: 490.05mg/dL (RR: 212–360mg/dL)].
Hematological and biochemical indices of proband and her daughter.
| Unit (SI) | Proband | Daughter | |
|---|---|---|---|
| White blood cell count | ×109/L | 7.4 | 6.9 |
| Red blood cell count | ×1012/L | 5.67 | 5.07 |
| Hemoglobin | g/L | 10.1 | 9.9 |
| Hematocrit | % | 33.3 | 33.8 |
| Mean corpuscular volume | fL | 58.7 | 66.7 |
| Mean corpuscular hemoglobin | Pg | 17.8 | 19.5 |
| Mean corpuscular hemoglobin concentration | g/dL | 30.3 | 29.3 |
| Platelet count | ×109/L | 169 | 171 |
| Serum creatinine | μmol/L | 0.05 | 0.08 |
| Aspartate transaminase | U/L | 12.7 | 15.3 |
| Alanine transaminase | U/L | 12.5 | 9.0 |
| Total bilirubin | μmol/L | 0.03 | 0.34 |
| Lactate dehydrogenase | U/L | 198 | 189 |
| Iron | μmol/L | 5.027 | 5.258 |
| Transferrin | g/L | 490.05 | 462.09 |
| Ferritin | pmol/L | 138.7 | 111.6 |
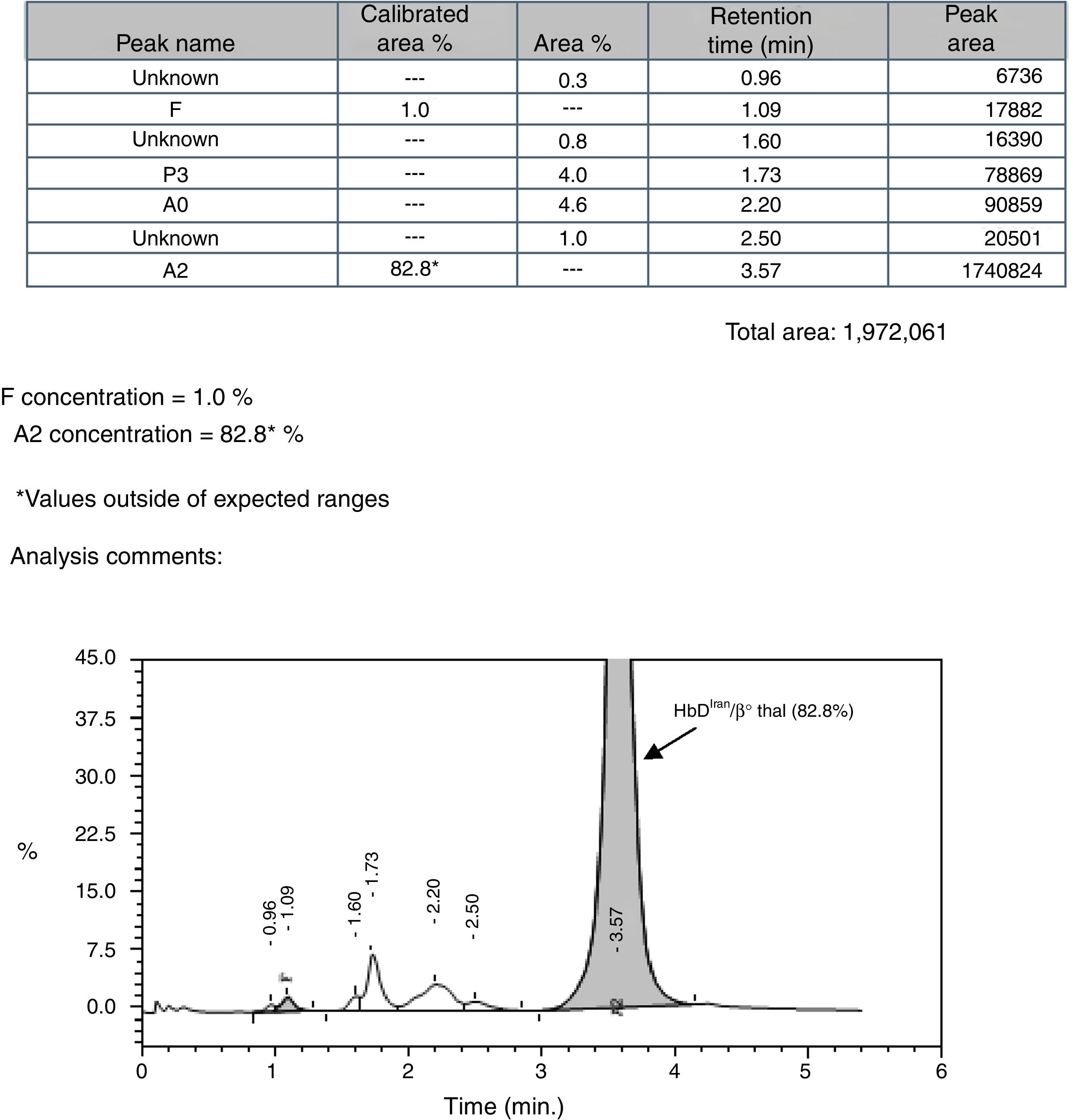
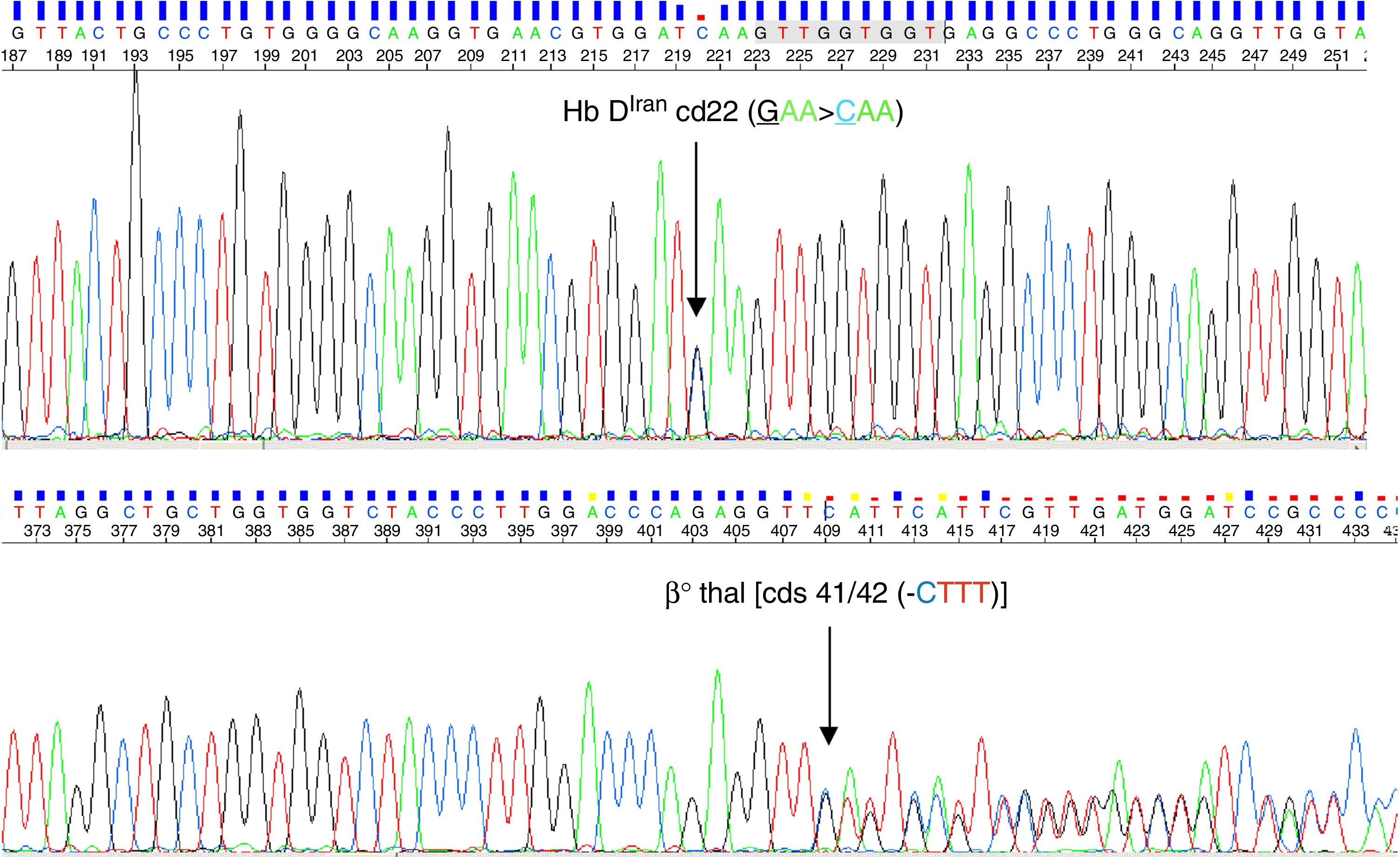
Because of the endemicity of the sickle cell hemoglobinopathy and its combination with β-thalassemia in this region, the sickling test and alkaline agarose gel Hb electrophoresis were performed; the sickling test was negative and a single band in the Hb S/D position was observed by Hb electrophoresis (pH-8.6). Cation exchange high performance liquid chromatography (CE-HPLC) was performed using the VARIANT-II hemoglobin testing system with the CDM 5.1.1™ software and Beta Thal Short Program (Bio-Rad Laboratories, Hercules, CA, USA) which showed a prominent peak in the Hb A2 window [82.8%; retention time (RT): 3.57min] with low Hb A0 and Hb F peaks (4.6% and 1.0%, respectively) (Figure 1). The possibility of homozygous Hb E was ruled out by the absence of a band in the position of Hb A2/Hb E by Hb electrophoresis. Hence, the case was initially suspected to be a rare finding of Hb Tianshui, which has similar alkaline Hb electrophoresis and CE-HPLC findings as reported earlier.3 However, the peak morphology characteristic of the present case was different from that of Hb Tianshui. Consequently, β-globin gene sequencing using the Big-Dye terminator protocol Ver 3.1 in an automated ABI-3730 DNA Analyzer (Applied Biosystems, USA) confirmed the case to be compound heterozygote of Hb DIran (β22 (B4) Glu>Gln; HBB: c.67G>C, GAA>CAA, rs33959855) (Figure 2A) with a β0-thalassemia mutation [4-base pair (bp) deletion at cds 41/42 (-CTTT)] (Figure 2B). An additional investigation for the XmnI polymorphism and deletional alpha thalassemia revealed that the proband was a homozygote for the wild type allele (CC) in the XmnI locus and heterozygous for the 3.7kb deletional alpha thalassemia. The presence of the homozygous wild allele in the XmnI locus corroborates the observed low Hb F level in this case, while the possible effect of heterozygous deletional alpha thalassemia on the microcytic hypochromic red cell parameters of the case could be masked by the simultaneous presence of the cds 41/42 (-CTTT) mutation.
Discussion![CE-HPLC showing characteristic peak of HbDIran/β0 thal [cds 41/42 (-CTTT)].](https://static.elsevier.es/multimedia/25311379/0000004000000001/v2_201911300802/S1516848417301226/v2_201911300802/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92xcqUtYYCc6yiYRjTD2AR3zJSEmH4GGT8A27de8H5QqAdccHlDwud9PYwTAyLTEDmW2DnpSRRGACrkCATql/9ED1BTPYOQt3tx4Ezf6+UEQF/ASW7tU6o8lmFEw2zNG0B5wjsNTJA08BQ7EzHLSjsfU5sUWMRUILCRdMarux+iUrIX//Uj9UkU5aO2rhjT2JKRKZi0F/qWI9WCB3yvihxAr2WZYpV2gPUB4wCgLsHOSUhX5SFB4RgYYE/xXftcTM5id9Qf2mi/XoUV1u2ZcKW0ysdH/NXGX3Rtp6oAAHtrC "CE-HPLC showing characteristic peak of HbDIran/β0 thal [cds 41/42 (-CTTT)].")
 DNA sequence chromatogram showing HbDIran mutation on 22 codon (GAA>CAG). (B) DNA sequence chromatogram showing β0 thal {4 bp del Cds 41/42 (-CTTT)}")
The Hb DIran trait and homozygous cases have been reported earlier.4,5 However, few studies have reported compound heterozygotes of Hb DIran with other Hb variants like Hb S and Hb DPunjab, β+-thalassemia IVS1–5 (G>C), β0-thalassemia (619bp-deletion) and undefined β-thalassemia from India and Pakistan. Various studies have reported that the quantity of Hb DIran eluting in the Hb A2 window in HPLC varies from 36.0 to 47.7% in a heterozygous condition, while in compound heterozygous states, the quantity varies between 47.3 and 94.4% (with Hb DPunjab, Hb S, β-thalassemia with the 619bp deletion mutation and beta thalassemia with unknown mutation).6–10 Almost all these cases were mild in presentation with concomitant anemia.
Codon 22 (GAA), is a mutational hotspot in exon I of the human β globin gene, although it does not take part in α-β or protein-heme interactions, as this is an external residue positioned at the B4 site of the helix. To date, six Hb variants (Hb DIran, Hb E-Saskatoon, Hb G-Coushatta, Hb D-Granada, Hb G-Taipei and Hb Bury) and one β0-thalassemia mutation [Codon 22 (G>T); GAA(Glu)>TAA (stop codon)] have been reported involving this codon. In Hb DIran, the change of glutamate to glutamine leads to an overall change of charge from negative to positive resulting in a protein that migrates to the position of Hb S in alkaline Hb electrophoresis.1,10 This rare variant has heat stability with no effect on oxygen equilibrium, intracellular 2,3-diphosphoglycerate or the Bohr effect.10 The homozygous state of Hb DIran reveals a milder phenotype even when Hb DIran co-inherits with β0-thalassemia.5,9 The present case agrees with this as evidence from the clinical and hematological investigations show. Although Hb DIran in combination with β-thalassemia produces a moderate microcytic and hypochromic red cell picture that is not transfusion dependent, the appearance of Hb DIran in the position of Hb S in alkaline agarose gel electrophoresis can lead to significant confusion and might falsely be reported as a sickle cell hemoglobinopathy unless a sickling test and HPLC are read together with these findings. Hb S can easily be distinguished from Hb DIran by performing CE-HPLC.
Reportedly in CE-HPLC, nine abnormal Hbs elute in the Hb A2 window (3.27–3.83 as per the manufacturer's guidelines in the operating software): Hb Deer Lodge, Hb Lepore, Hb DIran, Hb E, Hb Hamadan, Hb Osu-Christiansborg, Hb Tianshu, Hb G Honolulu and Hb G Copenhagen. Among these, Hb Deer Lodge, Hb Lepore and Hb DIran elute prior to the standard RT of Hb A2 (3.6min) while others have higher RT to that of Hb A2. Interestingly, Hb Lepore has the lowest average quantity (7–15%) followed by Hb G Honolulu (about 15% of total hemoglobin quantity) and Hb E (about 30% of total hemoglobin in absence of α-thalassemias). All the other variants eluting in the Hb A2 window have variant hemoglobin quantities higher than 30% on average under heterozygous conditions, making it difficult to distinguish in HPLC. Amongst these, Hb DIran has been reported to elute in this window at a RT of 3.49–3.58min; almost in the middle of the window (3.27–3.83). However, the pattern of mobility of these variants in alkaline electrophoresis is quite interesting. Hb E stands apart as its mobility is at the position of Hb A2 and can be easily identified. Hb Deer Lodge and Hb G Copenhagen have almost similar alkaline electrophoretic mobility i.e., slightly anodic to the position of Hb S. The rest of the variants are very difficult to distinguish even in alkaline electrophoresis because of their identical mobility to the position of Hb S. Additionally, all of these variants have a negative sickling test result.11
Further, as Hb DIran elutes in the Hb A2 window in HPLC masking elevated Hb A2, it becomes difficult to suspect the presence of β-thalassemia and direct gene sequencing needs to be performed. To the best of our knowledge, this is the first report of Hb DIran with β0-thalassemia [cds 41/42 (-CTTT)] reported from Odisha, India.
FundingThe study was performed under the Odisha Sickle Cell Project, funded by the National Health Mission of India, Odisha, India.
Conflicts of interestThe authors declare no conflicts of interest.
We acknowledge the support from Prof. Bijaya Kumar Dutta, Dean & Principal, Veer Surendra Sai Institute of Medical Science and Research (VIMSAR), Burla, Odisha. The authors are indebted to late Dr. Dilip Kumar Patel, Ex-Associate Professor, Department of Medicine, Veer Surendra Sai Medical College (now VIMSAR), Burla, Sambalpur, Odisha and Ex-Project Coordinator, Odisha Sickle Cell Project (NHM, Odisha) for his inestimable contribution to this study. The authors acknowledge the support of the Director, Anthropological Survey of India, Ministry of Culture, Government of India for sanctioning the collaboration program (Vide letter No. 18-22/PMI/2011, dated June 15 2013).


