



El aseguramiento de la calidad de la fase preanalítica se orienta hacia 2 aspectos clave: la gestión de los errores preanalíticos desde la perspectiva de la seguridad del paciente, y la mejora y armonización de los procedimientos, basada en la aplicación de normativa además de recomendaciones profesionales. Al igual que el resto de las fases, debe incluir un programa interno de aseguramiento y la participación en programas de intercomparación entre laboratorios.
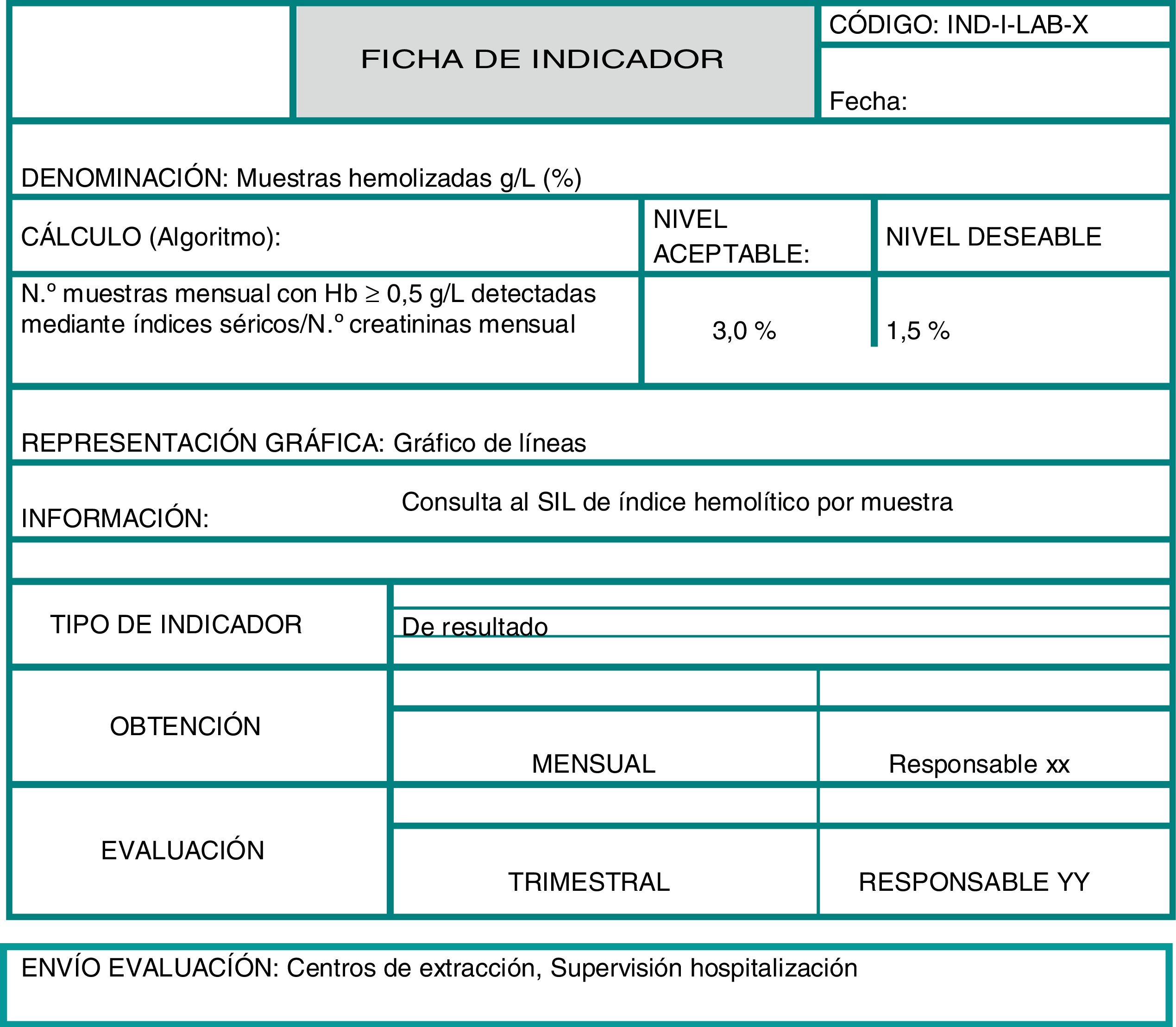
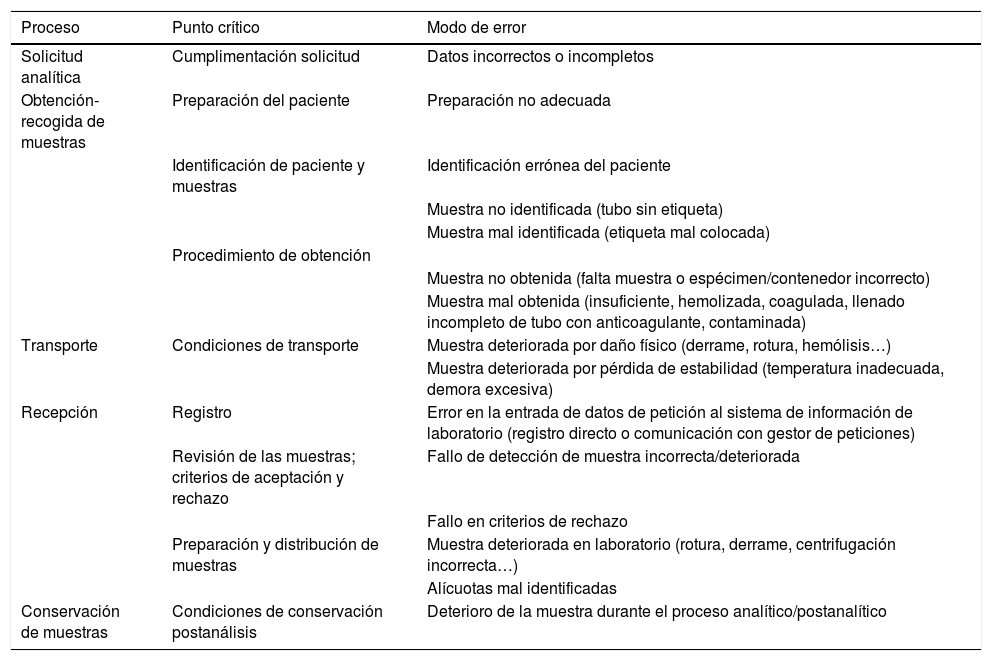
El control de calidad interno debe basarse fundamentalmente en la identificación de riesgos, detección sistemática de errores y establecimiento de indicadores. La selección de los indicadores priorizando el impacto en el paciente, la forma de detectar y registrar los errores de forma sistemática y fácilmente explotable, así como las variables utilizadas en su cálculo, son aspectos importantes para medir la eficacia de las acciones de mejora y permitir la comparabilidad entre laboratorios. En este sentido, los programas externos de la calidad de la fase preanalítica basados en la comparación de indicadores, son una herramienta útil para el diseño e implantación de un programa de aseguramiento de la calidad.
Este documento pretende servir de apoyo para que cada laboratorio seleccione, implante y evalúe sus propios indicadores, de acuerdo a las características individuales de sus procedimientos preanalíticos, pero sin perder de vista la armonización entre laboratorios.
The quality assurance of the pre-analytical phase is oriented towards two key aspects; the management of pre-analytical errors from the perspective of patient safety, and the improvement and harmonisation of procedures, based on the application of regulations and professional recommendations. Like the rest of the phases, it should include an internal quality assurance program, as well as the participation in external quality assurance programs.
The internal quality control should mainly be based on the identification of risks, systematic detection of errors, and establishment of indicators. The selection of indicators prioritising the impact on the patient, the way to detect and record errors in a systematic and easily exploitable manner, and also the variables used in the calculations, are important aspects to measure the effectiveness of improvement actions and to allow comparability between laboratories. In this sense, the external quality assurance programs of the pre-analytical phase based on the comparison of indicators are a useful tool for the design and implementation of a quality assurance program.
This document is intended as a support for each laboratory to select, implement, and evaluate its own indicators, according to the individual characteristics of its pre-analytical procedures, but without losing sight of the harmonisation between laboratories.
Artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
