



La mucolipidosis tipo II alfa/beta, también conocida como I-cell disease (MIM 252500), es una enfermedad metabólica consistente en una alteración en el tráfico de las hidrolasas lisosomales, causada por la actividad deficiente de la N-acetilglucosaminil 1-fosfo (NAcGlc-1-P) transferasa, responsable del paso inicial en la generación del marcador molecular manosa-6-fosfato. NAcGlc-1-P-transferasa es una enzima multimérica compuesta de tres subunidades polipeptídicas codificadas por dos genes diferentes (α, β y γ), el gen que codifica las subunidades α y β (GNPTAB), localizado en el cromosoma 12q23.3, se encuentra alterado en MLII. Hasta la fecha, han sido descritas al menos 20 mutaciones missense/nonsense en GNPTAB relacionadas con esta enfermedad autosómica recesiva. En este estudio, caracterizamos las alteraciones moleculares de un nuevo caso de mucolipidosis II, que presentaba importantes defectos esqueléticos.
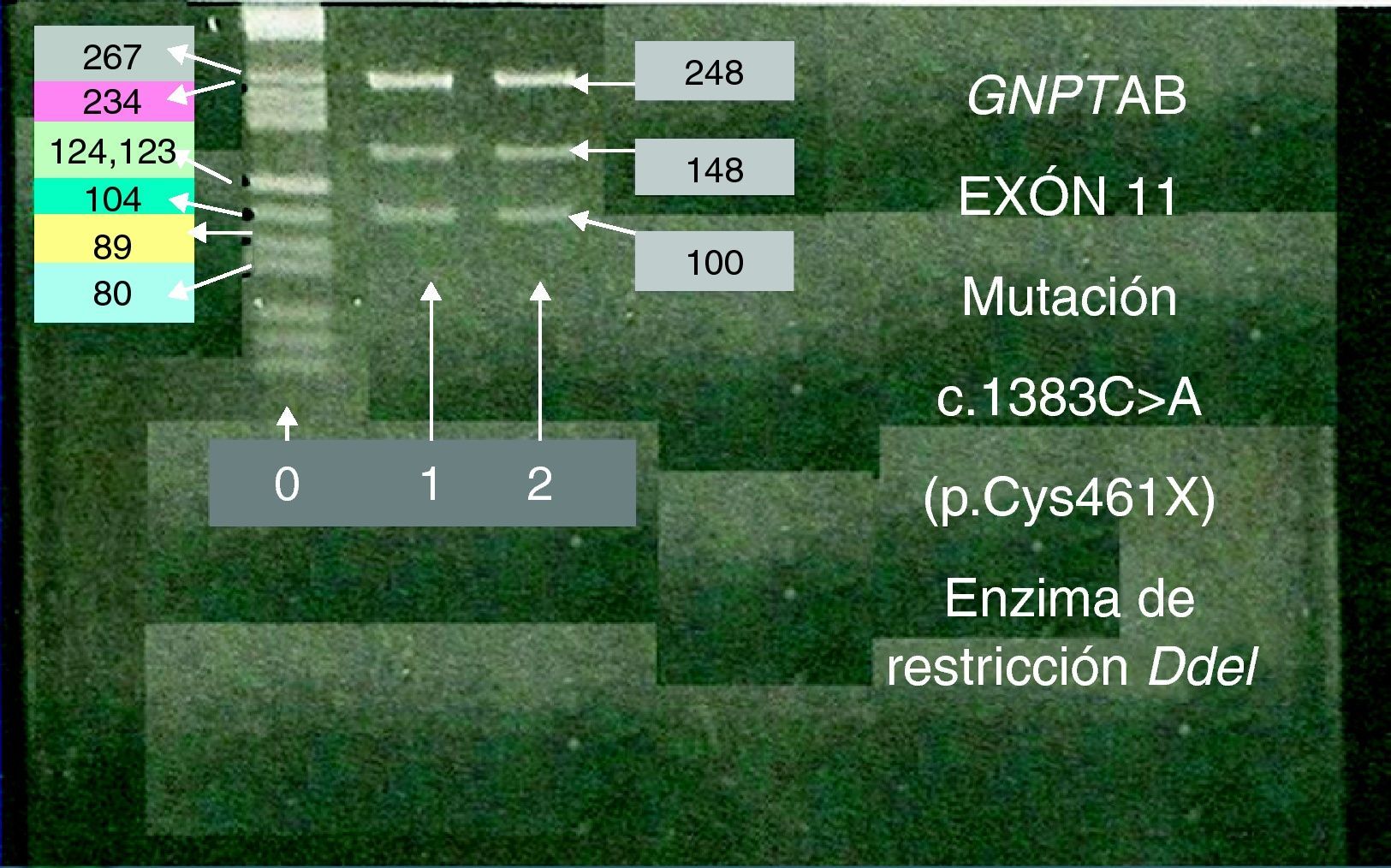
Materiales y métodosDeterminación de la actividad de las hidrolasas lisosomales en fibroblastos del paciente, plasma y medio de cultivo de los fibroblastos, mediante los correspondientes sustratos fluorogénicos. Secuenciación de todos los exones y de las regiones intrón-exón del gen GNPTAB, tras la amplificación del DNA genómico del paciente. Además, se analizó el exón 11 de todos los familiares por enzimas de restricción.
ResultadosLa actividad residual hallada de las hidrolasas lisosomales en los fibroblastos del paciente fue de aproximadamente 14% frente a los controles, mientras que la actividad de estas enzimas se multiplicó por 32 en plasma y por 9 en el líquido extra celular de los fibroblastos en cultivo del paciente, con respecto a los valores normales. Se identificaron dos nuevas mutaciones nonsense en GNPTAB asociadas con MLII, c.1383C>A (p.Cys461X) y c.3410T>A (p.Leu1136X), para las que el paciente fue heterocigoto compuesto.
ConclusionesCaracterización de los defectos moleculares de GNPTAB en un nuevo caso de MLII y la identificación de dos mutaciones noveles sin sentido, que facilitarán el diagnóstico prenatal de la enfermedad en la familia del paciente.
Mucolipidosis type II alpha/beta (MLII or I-cell disease) (MIM 252500) is a rare inborn lysosomal hydrolase trafficking disorder caused by the deficient activity of N-acetylglucosaminyl 1-phospho (NAcGlc-1-P) transferase, the enzyme responsible for the initial step in the generation of the mannose 6-phosphate recognition marker. NAcGlc-1-P-transferase is a multimeric enzyme composed of 3 polypeptide subunits (α, β and γ) encoded by 2 different genes. The gene encoding for the α/β subunits (GNPTAB), located on chromosome 12q23.3, is altered in MLII. To date, at least 20 missense/nonsense GNPTAB mutations have been described and incriminated in this autosomal recessive disorder. In this study, we characterized the molecular defect of a new case of MLII, presenting important skeletal abnormalities.
Material and methodsThe activity of lysosomal hydrolases in the patient's fibroblasts, plasma and cell culture medium was determined using appropriate fluorogenic substrates. All exons, as well as exon-intron boundaries, of the GNPTAB gene were sequenced after PCR amplification of the patient's genomic DNA. GNPTAB exon 11 was also studied by enzyme restriction analysis in the whole family.
ResultsIn the patient's fibroblasts, a residual activity of lysosomal hydrolases averaging 14% of control values was found, while a 32 and 9-fold increase in the activity of these enzymes was detected in plasma and the fibroblast culture medium, respectively. Two novel nonsense disease-associated GNPTAB mutations, c.1383C>A (p.Cys461X) and c.3410T>A (p.Leu1136X) were identified, the patient being a compound heterozygote.
ConclusionsCharacterization of the GNPTAB molecular defects in a new case of MLII and the identification of two novel nonsense mutations facilitated the prenatal diagnosis of this disease in the patient's family.
Artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
