



La neuroacantocitosis es una entidad que engloba diferentes enfermedades neurodegenerativas en las que, además de la sintomatología neurológica, se observan eritrocitos anormalmente espiculados. Dentro de este grupo se encuentra la coreoacantocitosis, una enfermedad autosómica recesiva que suele iniciarse en la segunda década de la vida1. Cursa, como clínica más característica, con comportamientos obsesivos, trastornos del control de impulsos, infantilismo, deterioro cognitivo, convulsiones, niveles elevados de CPK, distonía, corea y tics como síntomas más característicos2,3.
Esta enfermedad está asociada a mutaciones en la región 9p21.2 del gen VPS13A, que codifica la proteína coreina localizada en el cerebro, testículos, riñones, bazo y en los eritrocitos. El déficit de esta proteína genera una apoptosis de las neuronas estriatales, aunque su función real no está del todo dilucidada4,5.
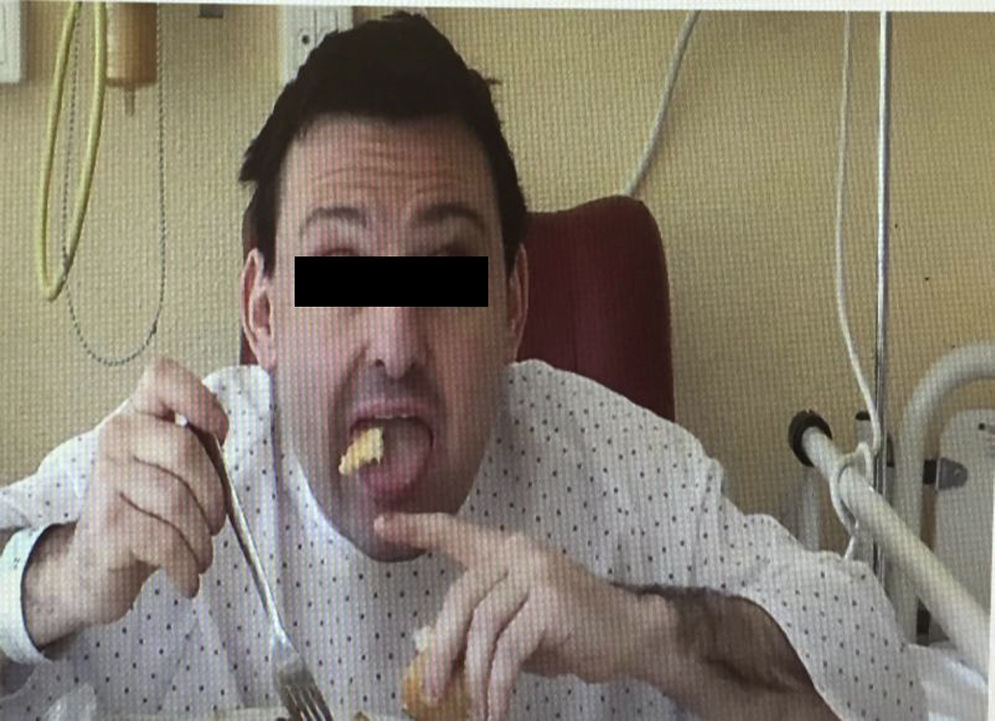
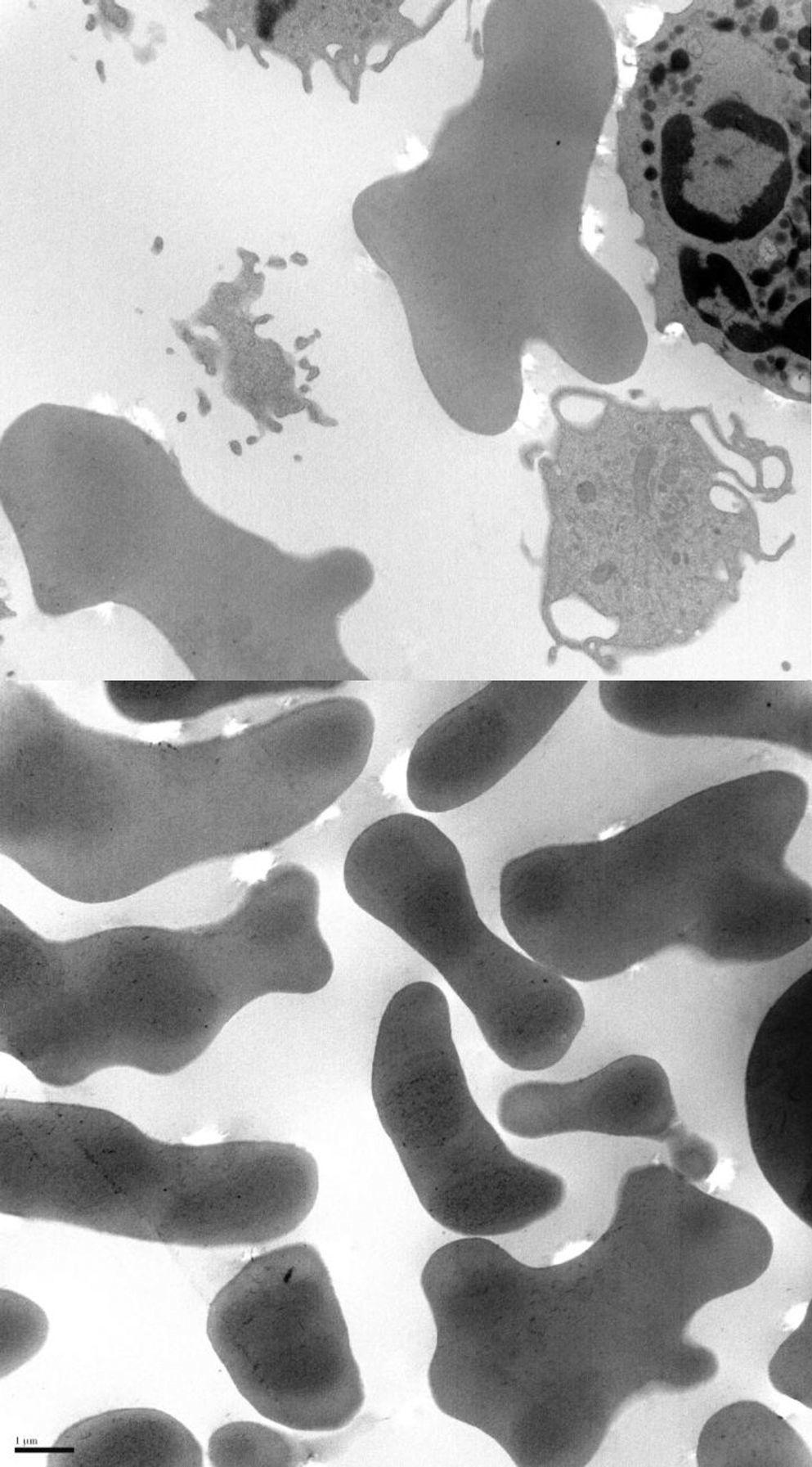
El paciente es un varón de 38 años con desarrollo psicomotor normal hasta la edad de 26 años en la que sufre su primera convulsión. No presentaba ningún antecedente personal ni familiar de interés, y hasta entonces no destacaba la toma crónica de fármacos. Tras las crisis mostraba valores de CPK muy elevados (superiores a 4.000Ul) que no llegaban a normalizarse en los periodos intercríticos (en torno a 600Ul). En la exploración presentaba una leve disartria, numerosos tics faciales y, durante la alimentación, sufría unos movimientos distónicos linguales que le desencadenaban la expulsión del bolo alimenticio (fig. 1). Se le practicó un examen neuropsicológico sin observarse deterioro cognitivo, si bien, mostraba un comportamiento infantil, cada vez más desinhibido e impulsivo. La neuroimagen constató una atrofia de la cabeza de los núcleos caudados y una discreta hiperlucencia en los putámenes. El antígeno de Kell fue negativo. La neurofisiología (EMG) fue normal. No se encontró mutación en el gen IT-15. Ante la presencia de crisis, CPK alta, tics y distonía lingual, asociado al hallazgo radiológico, se solicitó un frotis sanguíneo que mostró la presencia de escasos acantocitos, no suficientes como para considerarlo patológico. Dada la fuerte sospecha clínica, el servicio de hematología recurrió a la técnica de Feinberg, en la que se diluyen y cultivan los eritrocitos en suero salino o en EDTA lo que aumenta fuertemente el porcentaje de muestras positivas6. Con ello se observó que cuando se aumentaba el tiempo de incubación, también lo hacía el número de acantocitos hasta una tasa claramente patológica (fig. 2). Con la sospecha clínica de coreoacantocitosis se solicitó un estudio genético. En el estudio molecular se detectaron 2 mutaciones diferentes en heterocigosis en el gen VPS13A. Por una parte se detectó un cambio de una C por una G (c.1901-3C>G), que podría tratarse de un cambio patológico que afectase al correcto procesamiento del ARNm. En el otro alelo, se detectó una delección de 4 nucleótidos (c.9446_9449del) la cual produce, presumiblemente, un cambio en la pauta de lectura y la aparición de un codón de parada prematuro (p.ILe3149Thrfs*38). Estas alteraciones genéticas no habían sido descritas previamente en las bases de datos consultadas por nosotros, no obstante, lo más probable es que se tratase de un cambio con implicaciones patológicas. Se realizó el estudio genético de los progenitores, confirmándose que cada uno de ellos era portador de una de estas mutaciones. Con ello, se confirmaba que las alteraciones moleculares observadas en el paciente se situaban en diferentes alelos siendo muy verosímil que los cambios genéticos fuesen los responsables de la sintomatología neurológica.


La enfermedad tiene una gran variabilidad genética. Tomiyasu et al. en el 2011, encontraron 36 mutaciones patogénicas de las cuales hasta 20 no habían sido descritas previamente. En 16 de esos pacientes observaron una ausencia total de coreina en los eritrocitos7.
Para el control de las crisis se utilizaron diversos antiepilépticos con malos resultados. Desde su diagnóstico, se pautó tratamiento con fenitoína 150mg/8h (necesarios para conseguir niveles terapéuticos en sangre) manteniéndose sin crisis desde entonces. Para la terapéutica de los trastornos del comportamiento y de las discinesias se pautó tratamiento con ISRS, clonazepam, tetrabenazina, trihexafenidilo, pimozida y risperidona sin haber conseguido ningún tipo de mejoría clínica.
Queremos resaltar que el estudio de acantocitos mediante frotis puede dar falsos negativos y, ante una sospecha clínica fuerte, debe pedirse al servicio de hematología que realice la técnica de Feinberg. Que el control de las crisis epilépticas fue muy bueno con fenitoína, nuestro absoluto fracaso en el control de los trastornos del movimiento y del comportamiento y, por último, la presencia de 2 mutaciones diferentes en cada alelo de nuestro paciente, nunca descritas previamente, que dan lugar a una manifestación florida de la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

