





La práctica clínica basada en la evidencia requiere la integración de la experiencia profesional individual con los mejores datos objetivos para tomar la mejor decisión terapéutica. Los datos científicos de mejor grado de evidencia derivan de ensayos clínicos controlados y aleatorizados e investigaciones de vigilancia farmacológica poscomercialización y metaanálisis. En muchas ocasiones durante nuestras actividades clínicas buscamos sin éxito el ensayo clínico que conteste a nuestras preguntas científicas. Es en estos momentos cuando en ocasiones nos planteamos la puesta en marcha de un ensayo clínico.
Si usted como investigador clínico tiene una pregunta científica (relevante), que potencialmente requeriría la realización de un Ensayo Clínico para alcanzar una respuesta y carece del respaldo de una Compañía Farmacéutica para llevarlo a cabo, quizá encuentre de utilidad la lectura de este artículo, en el que intentamos presentar de forma breve y clarificadora la normativa regulatoria para planificar un Ensayo Clínico, con la humilde intención de que se convierta en una herramienta útil para cualquier investigador independiente.
Evidence-based clinical practice requires integration of individual professional experience with the best objective data to make the best therapeutic decision. The best degree of scientific evidence derives from controlled, randomized clinical trials and post-marketing drug surveillance studies and meta-analyses. During our clinical activities, we often search unsuccessfully for a clinical trial which answers our scientific questions. It is at those times that we may sometimes consider the conduct of a clinical trial.
If you, as a clinical investigator, have a (relevant) scientific question that could potentially require the conduct of a clinical trial to achieve a response and have no support from a pharmaceutical company to perform it, you may find it useful to read this article, in which an attempt has been made to briefly and clearly explain the applicable regulations for planning a clinical trial. Our humble intention is that this publication becomes a useful tool for any independent researcher.
En el presente artículo intentamos presentar de forma breve y clarificadora la normativa regulatoria para planificar un Ensayo Clínico (EC), con la humilde intención de que se convierta en una herramienta útil para cualquier investigador independiente.
Comenzaremos señalando que es fundamental tener una pregunta científica importante, y es aconsejable que no seamos nosotros los únicos que creamos que la pregunta es relevante; sin duda, conversaciones con colegas y la revisión de la literatura nos ayudaran a concretar la calidad científica de esta pregunta.
Los términos Estudio Clínico o Ensayo Clínico definen toda investigación efectuada en seres humanos dirigida a determinar o verificar los efectos clínicos y/o farmacológicos, farmacodinámicos, reacciones adversas y/o estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el propósito de determinar su seguridad y/o eficacia1. Todos los EC con medicamentos que se realicen en España deben llevarse a cabo de acuerdo con las normas de Buena Práctica Clínica2 (BPC), una serie de requisitos que deben cumplirse y que garantizan la protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo, así como la fiabilidad de los resultados del mismo. Antes de continuar es importante conocer las diferentes fases de un EC. Los ensayos de fase I determinan la seguridad de un nuevo tratamiento; el objetivo de un ensayo de fase I es determinar la dosis más segura de un nuevo medicamento que los pacientes puedan recibir sin causar efectos secundarios adversos.
El principal objetivo de un ensayo de fase II es determinar la eficacia del nuevo tratamiento para combatir una enfermedad determinada. Si la comprobación preliminar de seguridad en la fase I ha sido satisfactoria, se pasa a esta fase, la cual involucra la administración del fármaco a individuos que presentan la enfermedad para la que se ha concebido su empleo.
El objetivo de un ensayo de fase III es comparar el nuevo tratamiento con el tratamiento estándar. Se trata de averiguar si el nuevo tratamiento es mejor, igual o menos eficaz que el tratamiento habitual. Aparte de verificar la eficacia del medicamento, se busca determinar manifestaciones de toxicidad previamente no detectadas. También conocidos como estudios de farmacovigilancia, los ensayos de fase IV consisten en el seguimiento del fármaco después de que ha sido comercializado. Se busca básicamente la detección de toxicidad previamente insospechada, así como de la evaluación de la eficacia a largo plazo. En cualquier caso, el marco legal vigente nos remite a la consulta de documentos legales como:
- -
LEY 29/2006, de 26 de julio3.
- -
RD 2183/2004, de 12 de noviembre4.
- -
ORDEN SCO/256/2007, de 5 de febrero5.
- -
Volumen X Eudralex – Clinical trials (desarrollo de la Directiva 2001/20/CE)6.
- -
Aclaraciones sobre la aplicación del RD 223/20047.
Un punto importante a tener en cuenta es escribir el protocolo en colaboración con los diferentes Servicios que van a participar en el desarrollo del mismo.
Es recomendable diseñar un calendario para desarrollar todas las tareas administrativas que le proporcionarán las autorizaciones que serán imprescindibles para que usted pueda iniciar el ensayo: aprobación por parte de los Comités Éticos de Investigación Clínica (CEIC) y Agencia Española de Medicamentos y Productos Sanitarios (AEMPS).
Es importante señalar que como investigador clínico “no se encuentra solo”. Es muy probable que su hospital, a través de la Unidad de Investigación, su Consejería de Salud, su Fundación, su CEIC y sin duda la AEMPS (tiene una Oficina de Apoyo a la Investigación Independiente) tengan personal con conocimiento y experiencia que podrán orientarle durante este proceso arduo y trabajoso de llevar a cabo todos los trámites éticos/legales y administrativos para poner en marcha un estudio clínico (tabla 1) y la elaboración de documentos esenciales antes, durante y después del inicio del ensayo (tablas 2, 3 y 4, respectivamente).
Puntos clave en el proceso de legalización de un Ensayo clínico
| Documentación |
| Escritura de protocolo |
| Diseño cuaderno de recogida de datos |
| Redacción del consentimiento informado |
| Preparacion archivo Promotor |
| Preparacion archivo Investigador |
| Solicitud del número EudraCT |
| Identificación de Centros participantes |
| Adquisición de material |
| Tramitación aprobacion CEIC |
| Preparación y envío dossier general CEIC |
| Respuesta aclaraciones CEIC |
| TRAMITACIÓN APROBACION AGEMED |
| Preparación dossier general AEMPS |
| Envío telemático AEMPS |
| Respuesta aclaraciones AEMPS |
| Seguro |
| Negociación y firma de contratos |
| Medicación |
| Selección de equipo monitorización externa |
| Manejo de datos/bioestadística |
| Inclusión pacientes |
| Comunicación cierre de centro a CEIC |
| Comunicación fin de estudio a AEMPS |
| Elaboracion de informe final |
| Publicación |
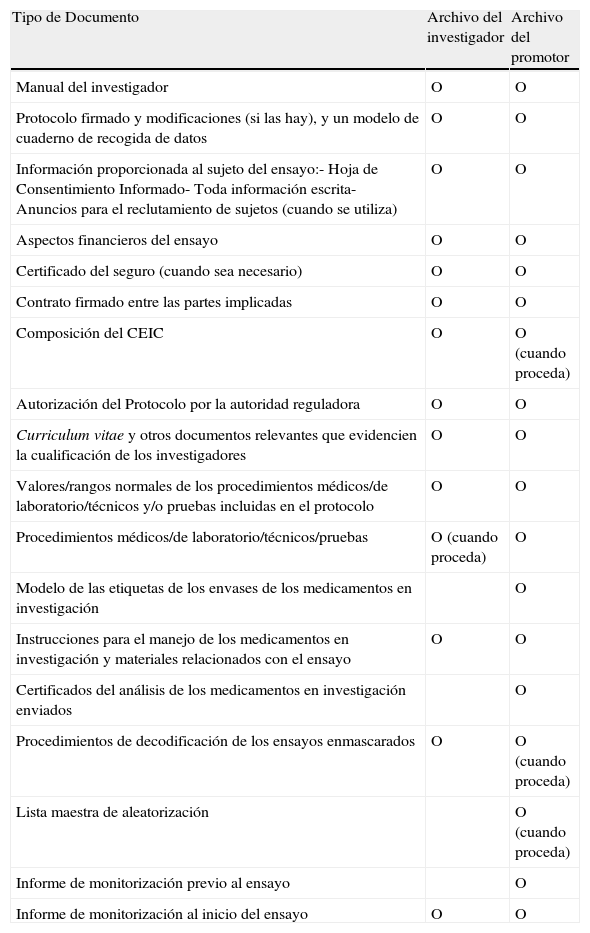
Documentos antes de iniciar el ensayo
| Tipo de Documento | Archivo del investigador | Archivo del promotor |
| Manual del investigador | O | O |
| Protocolo firmado y modificaciones (si las hay), y un modelo de cuaderno de recogida de datos | O | O |
| Información proporcionada al sujeto del ensayo:- Hoja de Consentimiento Informado- Toda información escrita- Anuncios para el reclutamiento de sujetos (cuando se utiliza) | O | O |
| Aspectos financieros del ensayo | O | O |
| Certificado del seguro (cuando sea necesario) | O | O |
| Contrato firmado entre las partes implicadas | O | O |
| Composición del CEIC | O | O (cuando proceda) |
| Autorización del Protocolo por la autoridad reguladora | O | O |
| Curriculum vitae y otros documentos relevantes que evidencien la cualificación de los investigadores | O | O |
| Valores/rangos normales de los procedimientos médicos/de laboratorio/técnicos y/o pruebas incluidas en el protocolo | O | O |
| Procedimientos médicos/de laboratorio/técnicos/pruebas | O (cuando proceda) | O |
| Modelo de las etiquetas de los envases de los medicamentos en investigación | O | |
| Instrucciones para el manejo de los medicamentos en investigación y materiales relacionados con el ensayo | O | O |
| Certificados del análisis de los medicamentos en investigación enviados | O | |
| Procedimientos de decodificación de los ensayos enmascarados | O | O (cuando proceda) |
| Lista maestra de aleatorización | O (cuando proceda) | |
| Informe de monitorización previo al ensayo | O | |
| Informe de monitorización al inicio del ensayo | O | O |
Documentos a archivar durante la realización del ensayo (1)
| Tipo de Documento | Archivo investigador | Archivo promotor |
| Actualizaciones del manual del investigador | O | O |
| Cualquier revisión a:- Protocolo, modificaciones y CRD- Documento de Consentimiento Informado- Toda información escrita facilitada a los sujetos- Anuncio para el reclutamiento de sujetos. (si se utiliza) | O | O |
| Autorización de la Agencia Reguladora de las modificaciones del protocolo y otros documentos | O (cuando proceda) | O |
| Curriculum vitae de nuevos investigadores | O | O |
| Actualizaciones de los valores/rangos normales de los procedimientos médicos/de laboratorio/técnicos y/o pruebas incluidas en el protocolo | O | O |
| Actualizaciones de los procedimientos Médicos/de laboratorio/técnicos/pruebas | O (cuando proceda) | O |
| Documentación de envío de los medicamentos en investigación y materiales relacionados con el ensayo | O | O |
| Certificados de análisis de los nuevos lotes de medicamentos en investigación | O | |
| Informes de las visitas de monitorización | O | |
| Comunicaciones relevantes diferentes a las visitas a los centros | O | O |
| Consentimientos informados firmados | O | |
| Cuadernos de recogida de datos completos, firmados y fechados | O (copia) | O (original) |
| Documentación de las correcciones en los CRD | O (copia) | O (original) |
| Notificación del investigador al promotor de los acontecimientos adversos graves y los informes relacionados | O | O |
| Notificación del promotor y/o investigador a la autoridad reguladora y los CEIC de las reacciones adversas graves e inesperadas y de otra información de seguridad | O (cuando proceda) | O |
| Notificación del promotor a los investigadores de información de seguridad | O | O |
| Informes intermedios o anuales al CEIC y autoridad reguladora | O | O (cuando proceda) |
| Registro de selección de sujetos | O | O (cuando proceda) |
| Lista de códigos de identificación de sujetos | O | |
| Registro de inclusión de sujetos | O | |
| Contabilización de los medicamentos en investigación en el centro del ensayo | O | O |
| Hoja de firmas | O | O |
| Registros de muestras de fluidos corporales/tejidos (si los hay) | O | O |
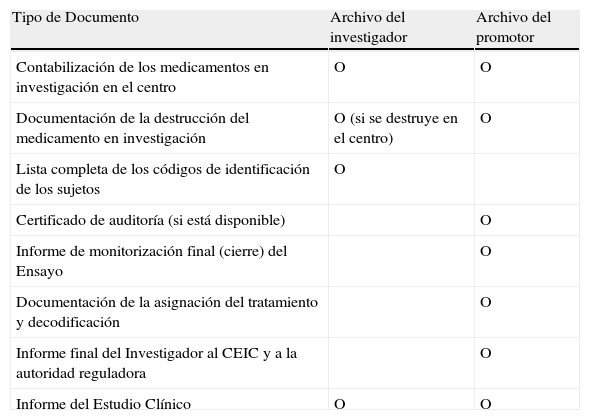
Documentos a archivar después de finalizar el ensayo
| Tipo de Documento | Archivo del investigador | Archivo del promotor |
| Contabilización de los medicamentos en investigación en el centro | O | O |
| Documentación de la destrucción del medicamento en investigación | O (si se destruye en el centro) | O |
| Lista completa de los códigos de identificación de los sujetos | O | |
| Certificado de auditoría (si está disponible) | O | |
| Informe de monitorización final (cierre) del Ensayo | O | |
| Documentación de la asignación del tratamiento y decodificación | O | |
| Informe final del Investigador al CEIC y a la autoridad reguladora | O | |
| Informe del Estudio Clínico | O | O |
La creación y puesta en marcha del Consorcio de Apoyo a la Investigación Biomédica en Red -CAIBER– (http://www.caiber.net) también aporta una infraestructura fundamental de apoyo a la investigación clínica independiente apoyando la puesta en marcha de EC aleatorizados prospectivos multicéntricos y de calidad.
Estos son algunos puntos clave que hemos trabajado en los últimos dos años para, primero, diseñar el estudio; segundo, obtener la aprobación de las autoridades regulatorias Europeas en materia de EC y, tercero, poner en marcha el Estudio TRAMOMTANA: TRATAMIENTO Multidisciplinar de la Obesidad Mórbida8.
DocumentaciónRedacción del Protocolo9En nuestro caso seguimos la Estructura Estándar de un Protocolo de EC con Medicamentos que pone a disposición de todos los investigadores la AEMPS en su página web, http://www.aemps.es/invClinica/estruEstandarProto.htm, detallada en la figura 1.
Diseño del cuaderno de recogida de datos.")
El diseño del cuestionario de recogida de datos es una fase crucial de cuya calidad dependerá en buena medida la calidad de los resultados finalmente obtenidos. Se debe hacer el máximo esfuerzo en seleccionar todos los datos que son relevantes para la hipótesis en estudio. No solo es imprescindible definir bien qué preguntas se efectuarán y qué mediciones se van a registrar (y en qué unidades de medida), sino también cómo se plantean las preguntas de forma clara, cómo se clasifican las respuestas y en qué lugar del cuaderno serán ubicadas.
Redacción del Consentimiento InformadoEn su redacción, el investigador debe cumplir la legislación pertinente, las normas de BPC1 y los principios éticos que tienen su origen en la declaración de Helsinki10. Antes del inicio del Ensayo, el investigador deberá tener el dictamen favorable por escrito del CEIC, tal como indica el art. 17 del RD 223/20042. Los CEIC disponen de modelos en sus diferentes páginas Web, que sirven de guía para la redacción de los mismos.
Preparacion y mantenimiento de los archivos del promotor y del investigadorEl promotor de un EC es la persona física o jurídica que tiene interés en la realización del ensayo, incluyendo su organización, comienzo y financiación; igualmente es responsable de firmar las solicitudes de autorización dirigidas al CEIC o a la AEMPS.
El investigador deberá estar cualificado por su titulación, formación y experiencia para responsabilizarse de la realización correcta del EC y deberá cumplir todos los requisitos especificados en la legislación pertinente.
Los archivos del promotor y del investigador incluirán una serie de documentos que detallamos en las tablas 2 (previo al inicio del EC), 3 (documentos a archivar durante la realización del ensayo) y 4, documentos a archivar una vez el ensayo ha finalizado.
Solicitud de número EudraCTCon el fin de proporcionar una identificación única para los EC en los que participe al menos un centro ubicado en la Comunidad, cada ensayo deberá identificarse por un número único (el número EudraCT11), que deberá constar en todas las solicitudes de EC que se presenten en algún Estado Miembro y se utilizará como elemento para identificar el ensayo en la documentación correspondiente (por ejemplo, en las notificaciones de reacciones adversas graves e inesperadas).
Identificación de centros participantesA la mayoría de los investigadores le gustaría que sus ideas se conviertiesen en relevantes proyectos multicéntricos, pero cuidado, nuestro mejor amigo puede no ser nuestro mejor socio.
Se debe comprobar que el investigador está cualificado por formación y experiencia, y tiene los recursos adecuados para realizar apropiadamente el ensayo, así como la idoneidad de las instalaciones del centro donde se pretende realizar dicho ensayo.
Adquisición de materialEl proceso de adquisición de material se verá beneficiado por una adecuada previsión. A la hora de presentar una solicitud para obtener financiación de proyectos de investigación, el presupuestar las diferentes partidas adecuadamente nos va a garantizar la viabilidad económica del proyecto.
Tramitación de la aprobacion del CEICEn cuanto a los CEIC, su papel es de vital importancia. El CEIC tiene, como indica el RD 223/20042, las siguientes funciones:
- a.
Evaluar los aspectos metodológicos, éticos y legales de los EC que les sean remitidos, de conformidad con lo establecido en la sección 2 del capítulo IV.
- b.
Evaluar las modificaciones relevantes de los EC autorizados.
- c.
Realizar un seguimiento del ensayo, desde su inicio hasta la recepción del informe final.
Cada CEIC requiere una serie de documentos (protocolo completo, cuaderno de recogida de datos, consentimiento informado, hoja de información al paciente, compromiso de publicación de resultados, declaración de conflictos de interés, etc.) que deben aportarse para que sea aceptado e incluido a revisión por los miembros del Comité.
Respuesta a aclaraciones del CEICTras la revisión de la documentación el Comité envía un informe al investigador con los puntos que deben ser incluidos o modificados antes de poder ser aprobado.
Este proceso suele tardar entre 1 o 2 meses en función del tipo de preguntas formuladas, subsanaciones a realizar y documentos solicitados por el Comité. En algunos casos extremos y en función de las dificultades en obtener la aprobación definitiva del protocolo, puede ser útil el solicitar defender el estudio en su reunión para poder contestar de primera mano las dudas/preguntas que han surgido en el CEIC.
SeguroPreviamente al inicio de la investigación clínica se concertará un seguro que cubra los daños y perjuicios que como consecuencia del mismo pudieran resultar para la persona en que hubiere de realizarse.
El promotor de la investigación clínica es el responsable de la contratación de dicho seguro de responsabilidad civil y este cubrirá las responsabilidades del promotor, del investigador y sus colaboradores y del titular del hospital o centro donde la investigación clínica se realice.
Cuando por cualquier circunstancia el seguro no cubra enteramente los daños, el promotor de la investigación clínica, el investigador principal y el titular del hospital o centro donde se realice la investigación clínica, son solidariamente responsables, sin necesidad de que medie culpa, del daño que en su salud sufra el sujeto sometido al EC, así como de los perjuicios económicos que de dicho daño directamente se deriven, siempre y cuando este sea consecuencia del tratamiento con el producto objeto de la investigación clínica o de las medidas terapéuticas o diagnósticas que se adopten durante la realización del mismo.
El coste de este seguro varía en función de tipo de población, número de pacientes incluidos, duración del ensayo y medicamento/proceso terapéutico utilizado. En la mayoría de los casos supone un desembolso significativo (6.000-12.000€). Cualquier excepción a este requisito debe ser adecuadamente justificada y esta excepción deberá ser valorada y aprobada por el CEIC.
Tramitación de la aprobación de la Agencia Española de Medicamentos y Productos SanitariosLa AEMPS tiene como misión garantizar a la sociedad, desde la perspectiva de servicio público, la calidad, seguridad, eficacia y correcta información de los medicamentos y productos sanitarios en el más amplio sentido, desde su investigación hasta su utilización, en interés de la protección y promoción de la salud de las personas y de los animales.
Envío telemático de solicitud de Ensayo Clínico a la AEMPSEl Portal de Ensayos Clínicos con Medicamentos de uso humano del Ministerio de Sanidad, Política Social e Igualdad, permite la elaboración y presentación de una nueva solicitud de EC y de cualquier tipo de solicitud relacionada con un EC en trámite a la AEMPS.
Podremos completar los formularios oficiales de solicitudes, imprimirlos, archivarlos y realizar la presentación telemática de dichas solicitudes así como adjuntar la documentación necesaria.
Le recomendamos que inicie el proceso desde http://aemps.es/aplicaciones/usoHum/ensaClin/portal_ensaClinicos.htm#entrada donde encontrará una gran cantidad de información detallada que le servirá de enorme ayuda.
A través de la Oficina virtual de la AEMPS (http://aemps.es/aplicaciones/home.htm), usted podrá acceder al Portal de Ensayos Clínicos con Medicamentos https://sinaem4.agemed.es/ecm/paginaPresentacion.do que permite la presentación de la solicitud inicial, EC en trámite y modificación relevante referente a un EC en formato electrónico, con carácter oficial y además, en ella encontrará manuales de ayuda para utilizar la aplicación telemática.
Antes de finalizar el proceso de envío encontraremos una pantalla en la que figuran una serie de documentos que es obligatorio adjuntar: Protocolo, Manual del Investigador, Tasa de una solicitud de Ensayo Clínico, Dictamen del CEIC, hoja de Información para los sujetos del ensayo, correo de asignación del número de EudraCT, Ficha Técnica, etc. Le recomendamos que le dé al archivo en .pdf el nombre descriptivo del tipo de documento al que se refiere, ya que este será el que posteriormente se muestre en el acuse de recibo.
Respuesta a aclaraciones de la AEMPSLas respuestas a aclaraciones pueden ser de dos tipos, con o sin modificación, en función de si se modifica o no algún documento presentado en la solicitud inicial.
Negociación y firma de contratosLos aspectos financieros del ensayo deberán estar documentados en un contrato entre el promotor y el investigador/institución1.
La negociación y firma de contratos en investigación clínica es un tema candente y con gran repercusión, ya que los contratos de realización de ensayos entre promotores y centros sanitarios públicos, son muy numerosos al tiempo que, por el elemento público, suscitan mayor complejidad jurídica.
El contrato es un instrumento necesario para la investigación clínica con medicamentos objeto de debate, principalmente debido al sistema de contratación de EC con centros públicos, así como por las dificultades (prácticas y jurídicas) que representa para los laboratorios la falta de homogeneidad en el contenido de dichos contratos (que varían ostensiblemente según el centro sanitario de que se trate).
Muchos de los actores reclaman un modelo único de contrato. ¿Qué razones hay para ello? Principalmente, se trata de asegurar la presencia de ciertos contenidos mínimos en dichos contratos, lo cual redundará en una mayor seguridad jurídica. Al mismo tiempo se persigue reducir los plazos de negociación: aspecto este con indudable trascendencia financiera, especialmente si los ensayos están vinculados a la comercialización de un medicamento que está en periodo de validez de una patente. ¿Por qué?:
Porque los EC se desarrollan generalmente consumiendo tiempo de validez de la patente que tiene obviamente su prioridad ubicada en fases previas del desarrollo. Cualquier retraso en el desarrollo de los productos que ampara supone consumir parte del periodo de comercialización exclusiva que otorgan tales derechos de patente, con lo que se disminuye la posibilidad de amortizar los costes incurridos en el desarrollo del producto. Esta urgencia contrasta con las constricciones administrativas del centro sanitario público, sus estructuras burocratizadas y la sobrecarga de trabajo de los órganos involucrados. El RD 223/2004, de 6 de febrero, de EC2, impone dos plazos que pueden solaparse en parte, como son el de la emisión del dictamen por el CEIC y la aprobación del ensayo por la AEMPS. Estos plazos, de acuerdo con la “Encuesta implementación RD 223/2004. Resultados tras 12 meses y comparación con la situación tras 6 meses” realizada y publicada por AMIFE y MDS Pharma Services, ocupan una media de 204 días en el mejor de los casos y nos referimos a la industria, mediante la participación de CRO (Clinical Research Organizations), lo que supone un tiempo considerable.
La negociación del contrato y su firma por las partes conlleva entre 24 y 64 días adicionales. Ante la imposibilidad por parte del promotor de acortar los plazos de emisión del dictamen por el CEIC y de la autorización por la AEMPS, los esfuerzos de reducción de plazos han de concentrarse en el periodo de negociación del contrato.
MedicaciónLa custodia, conservación y dispensación de los medicamentos corresponderá exclusivamente a los servicios de Farmacia de los Hospitales o de las estructuras de Atención Primaria del Sistema Nacional de Salud para su dispensación en dichas instituciones, de acuerdo con el artículo 2.6 de la Ley 29/20063.
Como el Servicio de Farmacia tendrá que colaborar con su estudio ya sea en sus conversaciones con la AEMPS y/o con la fabricación, importación, enmascaramiento, dispensación y control de la medicación o sustancia, sería recomendable que este Servicio ya participase en la redacción del Protocolo desde el principio. Si no lo hace en ese momento de diseño del estudio, es probable que lo deba hacer a posteriori y debido a la ausencia de comunicación previa con el Servicio, se pueda retrasar la aprobación final del proyecto.
Manejo de datos/bioestadísticaLa ley 41/2002, de 14 de noviembre, básica reguladora de la Autonomía del paciente y de derechos u obligaciones en materia de información y documentación clínica12, la Ley Orgánica 15/1999, de 13 de diciembre de Protección de datos de carácter personal13 y el RD 223/2004, de 6 de febrero2, por el que se regulan los EC con medicamentos ofrecen el marco legal aplicable a la gestión y control de los datos obtenidos en un EC. Es muy importante que el promotor garantice la especificación en el protocolo u otro acuerdo por escrito que el investigador o institución permitirán el acceso directo a los datos o documentos fuente para la realización de la monitorización, la auditoría, la revisión por el CEIC, así como la inspección del ensayo por las autoridades sanitarias.
Es necesario un plan estadístico en el que deben describirse los métodos estadísticos que se utilizarán, incluyendo el calendario de todos los análisis intermedios planificados.
Inclusión de pacientesAntes del inicio del ensayo el investigador deberá tener el informe favorable por escrito del CEIC del documento de Consentimiento Informado y de cualquier otra información escrita que se entregue a los sujetos, de acuerdo con el art. 17 del RD 223/20042.
Además, para la inclusión de sujetos en un EC se deberá cumplir la legislación pertinente, Título I, art. 4 de la Ley 14/2007, de 3 de julio, de Investigación Biomédica14, Título II, Capítulo II, artículo 13 del mismo texto legal, normas de BPC1 y los principios éticos que emanan de la declaración de Helsinki9.
Los pacientes deberán cumplir los criterios de inclusión tal como se indica en el Protocolo y ninguno de los criterios de exclusión en el momento de su inclusión en el ensayo.
Redacción de informes a promotor y actualizaciones a las autoridades competentesDurante la ejecución de un Ensayo se deben realizar y presentar diferentes tipos de informes al CEIC, agencia reguladora y/o promotor, como notificaciones del Investigador al promotor de los acontecimientos adversos graves y los informes relacionados, actualizaciones de los valores/rangos normales de los procedimientos médicos/de laboratorio/técnicos y/o pruebas incluidas en el protocolo, informe anual a CEIC (la mayoría de los CEIC tienen un procedimiento normalizado de trabajo -PNT- a tal efecto), comunicaciones relevantes diferentes a las visitas a los centros y otros que deberá archivar en todos los casos, como hacemos constar en la tabla 3.
Una vez finalizado el Ensayo, la Comunicación de Fin de Estudio a la AEMPS y la elaboración del Informe Final pondrán colofón al proceso, y podremos publicar los resultados de nuestra actividad investigadora con total garantía de calidad científico-ética y jurídico-normativa.
AgradecimientosA CAIBER, ISCiii y AEMPS.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.