


El desarrollo en los últimos años de las denominadas tecnologías de secuenciación masiva permite actualmente obtener millones de secuencias de ADN a una velocidad sin precedentes y a un coste cada vez más reducido. Estas tecnologías están permitiendo la consecución de logros científicos trascendentales, con la identificación de nuevos genes y la resolución de las bases genéticas de enfermedades mendelianas a la cabeza. Su potencial ha permitido el desarrollo de nuevas aplicaciones y pruebas biológicas que van a revolucionar, en un futuro próximo, el diagnóstico postnatal y prenatal de enfermedades genéticas. En el presente artículo se ofrece una visión general de la tecnología y se examinan sus ventajas e inconvenientes respecto a métodos convencionales así como algunas de las principales estrategias, incluyendo métodos de estrategias de diagnóstico prenatal dirigidas a la detección de aneuploidías y síndromes de deleción/duplicación.
The development in the recent years of the so-called next generation sequencing technologies based on massive parallel methods currently allows the production of millions of DNA sequences at an unprecedented speed with an increasing reduced cost per nucleotide. These technologies are producing very significant scientific achievements, with the identification of new genes and the resolution of the genetic basis of Mendelian diseases at the forefront. The potential of this technology is being used to create new applications and biological tests that are soon going to revolutionise the pre- and postnatal diagnosis of genetic disorders. In this paper we provide a general overview of the technology, examining its advantages and disadvantages in comparison with conventional strategies, as well as some of the main applicactions, including prenatal diagnosis strategies aimed at detecting aneuploidies and deletion/duplication syndromes.
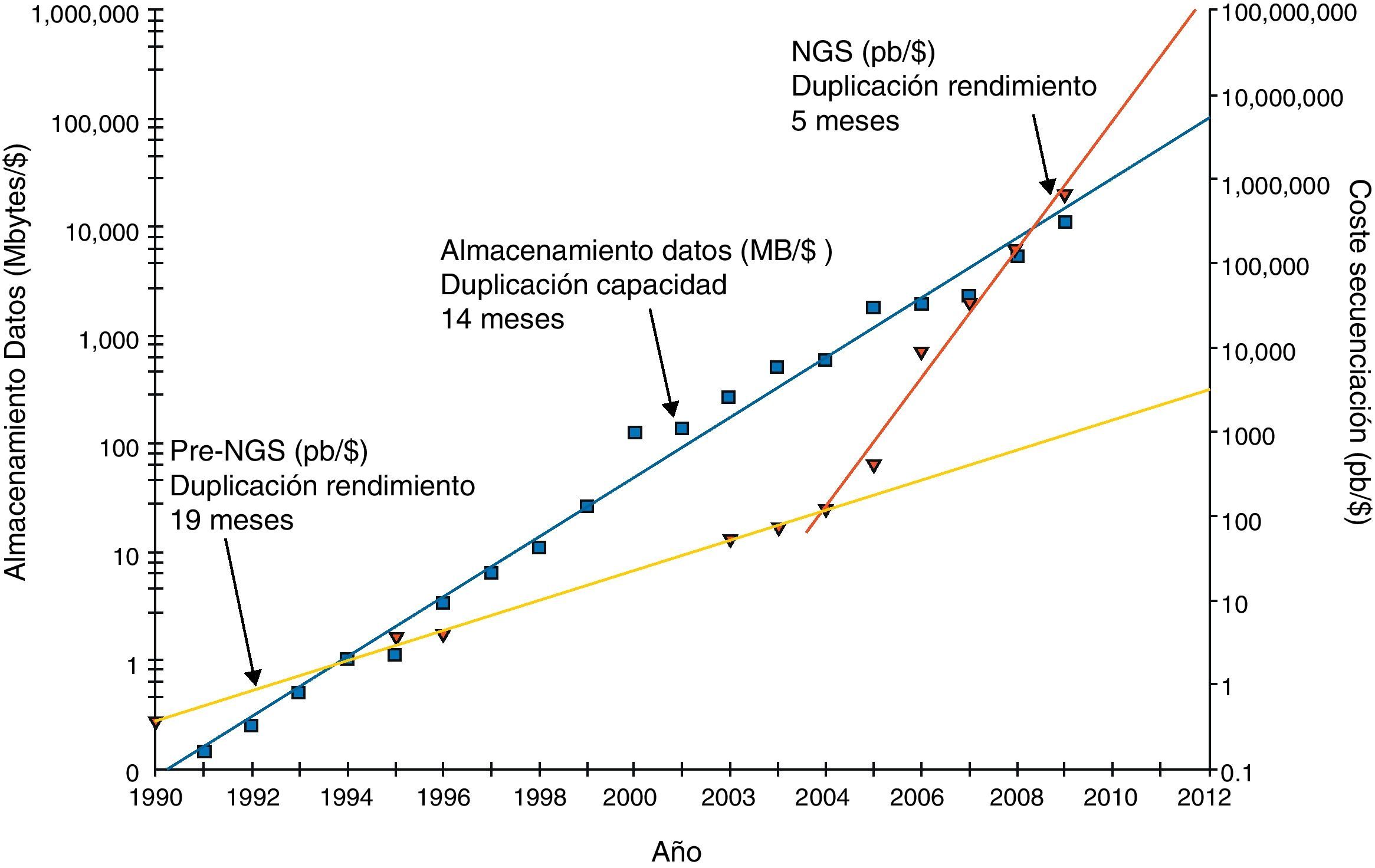
Aunque la tecnología convencional de secuenciación ideada por Sanger1 proporciona la resolución definitiva para detectar variantes genéticas de pequeño tamaño, tiene la limitación de solo poder realizar 96 o 384 reacciones en paralelo. Esto propicia que la ejecución de experimentos de secuenciación basados en esta técnica se prolongue mucho tiempo y que el precio por base secuenciada sea elevado (para grandes proyectos se estima un coste de 0,5US$ por kilobase [kb]; ∼1€ por cada 2,5kb)2–4. Los avances tecnológicos de los últimos 5 años han conducido al desarrollo de la secuenciación de nueva generación (next generation sequencing [NGS]), también conocida como secuenciación masiva paralela, del inglés massive parallel sequencing (MPS). Esta «nueva generación» ha mejorado dramáticamente en los últimos años, logrando que el número de bases que se pueden secuenciar por unidad de precio haya crecido exponencialmente (fig. 1)5. Por tanto las nuevas plataformas se distinguen por su capacidad de secuenciar millones de fragmentos de ADN de forma paralela a un precio mucho más barato por base (tabla 1). Además la secuenciación masiva tiene el potencial de detectar todos los tipos de variación genómica en un único experimento, incluyendo variantes de nucleótido único o mutaciones puntuales, pequeñas inserciones y deleciones, y también variantes estructurales tanto equilibradas (inversiones y traslocaciones) como desequilibradas (deleciones o duplicaciones).
 con un tiempo de duplicación en el rendimiento ligeramente inferior a la de almacenamiento hasta el año 2004, cuando la NGS produjo una inflexión de esta tendencia a menos de 6 meses (línea oblicua más corta situada a la derecha). Estos datos no están corregidos por la inflación o por los costes adicionales que incluirían costes de personal, depreciación y otros gastos generales ocasionados en los laboratorios. Esta figura es una adaptación tomada de una publicación previa (Stein, 2010)5.")
Historial de cambios en los precios de sistemas de almacenamiento y en los costes de las tecnologías de secuenciación.
$: dólar; MB: megabytes; NGS: next generation sequencing; pb: pares de bases.
Los cuadrados oscuros indican el coste de los discos de almacenamiento en megabytes por dólar estadounidense a lo largo de los años. La línea de tendencia oscura muestra el crecimiento exponencial de la capacidad almacenamiento por dólar con una tasa de duplicación aproximada de 1,5 años. El coste de la secuenciación expresado en pares de bases por dólar se puede observar gracias a los triángulos de la figura. Este valor muestra una tendencia exponencial (línea clara) con un tiempo de duplicación en el rendimiento ligeramente inferior a la de almacenamiento hasta el año 2004, cuando la NGS produjo una inflexión de esta tendencia a menos de 6 meses (línea oblicua más corta situada a la derecha). Estos datos no están corregidos por la inflación o por los costes adicionales que incluirían costes de personal, depreciación y otros gastos generales ocasionados en los laboratorios.
Esta figura es una adaptación tomada de una publicación previa (Stein, 2010)5.
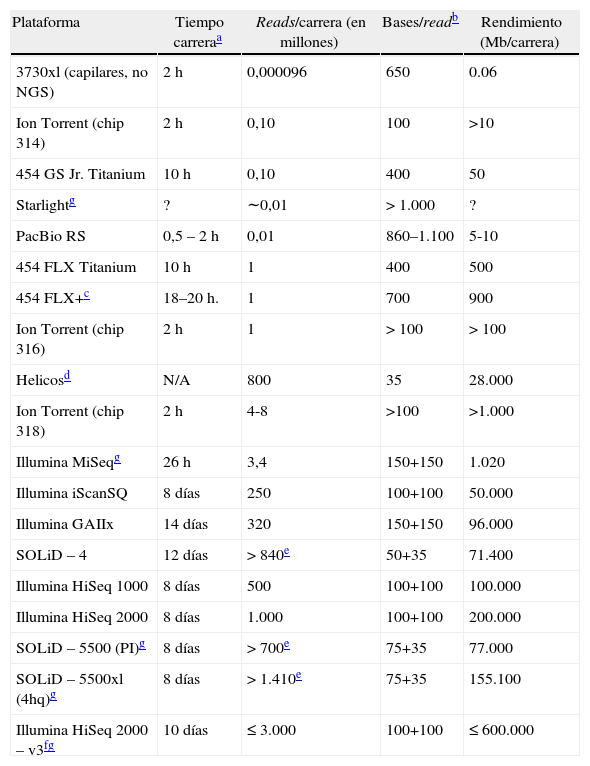
Comparativa entre diferentes plataformas de secuenciación
| Plataforma | Tiempo carreraa | Reads/carrera (en millones) | Bases/readb | Rendimiento (Mb/carrera) |
| 3730xl (capilares, no NGS) | 2 h | 0,000096 | 650 | 0.06 |
| Ion Torrent (chip 314) | 2 h | 0,10 | 100 | >10 |
| 454 GS Jr. Titanium | 10 h | 0,10 | 400 | 50 |
| Starlightg | ? | ∼0,01 | >1.000 | ? |
| PacBio RS | 0,5 – 2 h | 0,01 | 860–1.100 | 5-10 |
| 454 FLX Titanium | 10 h | 1 | 400 | 500 |
| 454 FLX+c | 18–20 h. | 1 | 700 | 900 |
| Ion Torrent (chip 316) | 2 h | 1 | >100 | >100 |
| Helicosd | N/A | 800 | 35 | 28.000 |
| Ion Torrent (chip 318) | 2 h | 4-8 | >100 | >1.000 |
| Illumina MiSeqg | 26 h | 3,4 | 150+150 | 1.020 |
| Illumina iScanSQ | 8 días | 250 | 100+100 | 50.000 |
| Illumina GAIIx | 14 días | 320 | 150+150 | 96.000 |
| SOLiD – 4 | 12 días | >840e | 50+35 | 71.400 |
| Illumina HiSeq 1000 | 8 días | 500 | 100+100 | 100.000 |
| Illumina HiSeq 2000 | 8 días | 1.000 | 100+100 | 200.000 |
| SOLiD – 5500 (PI)g | 8 días | >700e | 75+35 | 77.000 |
| SOLiD – 5500xl (4hq)g | 8 días | >1.410e | 75+35 | 155.100 |
| Illumina HiSeq 2000 – v3fg | 10 días | ≤3.000 | 100+100 | ≤600.000 |
h: horas; Mb: megabases.
∼Valor probablemente derivado de información no publicada indisponible en mayo de 2011.
Adaptado de Glenn, 201159.
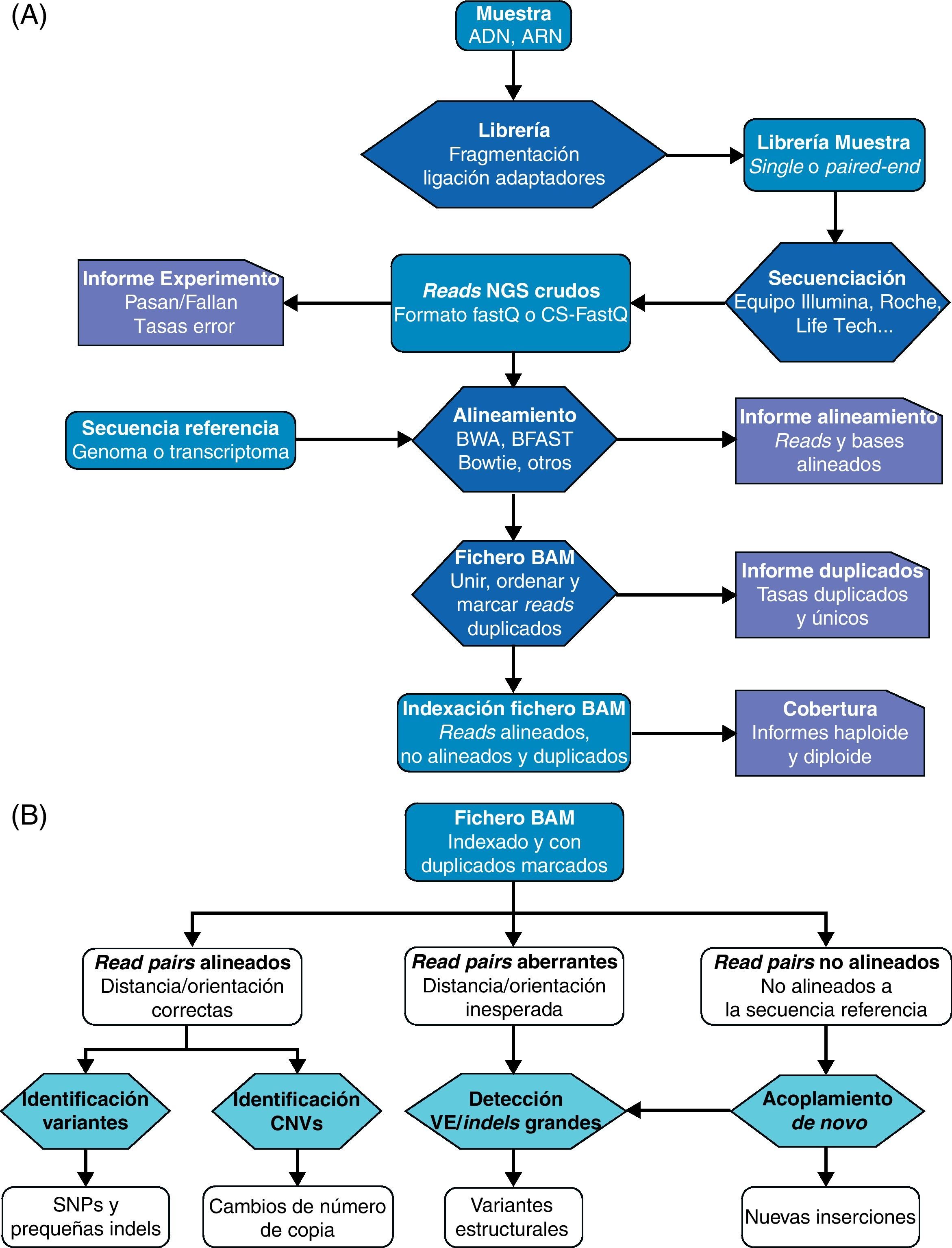
Las tecnologías de secuenciación implementadas en los distintos instrumentos actualmente utilizados para la NGS difieren en varios aspectos, pero el esquema principal de trabajo es conceptualmente similar para todos ellos6 (fig. 2A). El ADN se fragmenta y mediante ligación se le añaden secuencias adaptadoras a los extremos. Los fragmentos de ADN a continuación se amplifican clonalmente y se agrupan juntos (clustering) para ser utilizados como entidades a secuenciar. La secuenciación se realiza entonces alternando ciclos de terminación reversible cíclica (cyclic reversible termination [CRT]) y de toma de imágenes (imaging)6,7. La reacción CRT utiliza terminadores reversibles para incorporar nucleótidos marcados fluorescentemente que a continuación son «fotografiados» en la toma de imágenes y posteriormente son procesados. Las secuencias cortas producidas por el instrumento a partir de los extremos del ADN con los adaptadores se denominan lecturas o reads. En general, los nuevos secuenciadores generan lecturas a partir de cada uno de los extremos de un fragmento de ADN (el inserto), dando lugar a lecturas apareadas, y lo hacen usando dos estrategias diferentes. Los mate pairs se crean a partir de fragmentos de ADN de tamaño conocido (creando librerías con tamaños >600 pares de bases (pb) algunas librerías pueden alcanzar tamaños de inserto de 4kb), que se circularizan y se ligan usando un adaptador interno. Estos fragmentos circularizados se trocean al azar para luego purificar los segmentos que contienen el adaptador a partir del que se secuencia. Por contra, las lecturas de tipo paired end se generan mediante la fragmentación del ADN en pequeños segmentos (<300pb) de los cuales se secuencia el final de ambos extremos. Las lecturas paired end proporcionan rangos de tamaños de inserto más estrechos, mientras que las de tipo mate pair tienen la ventaja de cubrir tamaños mayores8,9. Un aspecto importante en la NGS es el número de veces que cada base del genoma está presente en los reads de secuenciación producidos. Este valor se denomina profundidad de cobertura (depth of coverage, o simplemente, coverage) y es uno de los factores determinantes para evaluar la fiabilidad del nucleótido asignado a esa posición del genoma. En experimentos de cuantificación de número de copias de variantes estructurales como deleciones y duplicaciones estos valores cobran una relevancia capital tal y como se discutirá más adelante.
 Secuenciación y alineamiento. Las librerías construidas con fragmentos de ADN o ARN se secuencian de forma masiva paralela. Tras la captura de imágenes y «traducción» a las secuencias de nucleótidos correspondientes, las lecturas o reads resultantes son alineados contra una secuencia de referencia. Los reads alineados y no alineados se exportan a un fichero de datos de tipo BAM o SAM y se marcan aquellos reads duplicados. B) Análisis del fichero de datos de alineamiento BAM. En el fichero BAM el estatus y calidad de alineamiento con la secuencia de referencia de cada read está indicado. Los pares de reads correctamente alineados se utilizan en la detección de SNP y pequeñas inserciones y deleciones y en la estimación del número de copias. Los reads alineados de forma aberrante, es decir, en los que los reads de un par muestran una distancia o una orientación inesperada, se analizan como indicadores potenciales de variantes estructurales. Por último, el acoplamiento de novo de reads no alineados con la referencia proporciona predicciones de variantes estructurales y de nuevas inserciones. Adaptado a partir de Koboldt et al.17. CNV: copy number variants (variantes variantes de número de copia); Indels: inserciones y deleciones; SNP: single nucleotide polymorphisms (polimorfismos de nucleótido único); VE: variantes estructurales.")
Esquema básico de trabajo en estudios de secuenciación masiva.
A) Secuenciación y alineamiento. Las librerías construidas con fragmentos de ADN o ARN se secuencian de forma masiva paralela. Tras la captura de imágenes y «traducción» a las secuencias de nucleótidos correspondientes, las lecturas o reads resultantes son alineados contra una secuencia de referencia. Los reads alineados y no alineados se exportan a un fichero de datos de tipo BAM o SAM y se marcan aquellos reads duplicados. B) Análisis del fichero de datos de alineamiento BAM. En el fichero BAM el estatus y calidad de alineamiento con la secuencia de referencia de cada read está indicado. Los pares de reads correctamente alineados se utilizan en la detección de SNP y pequeñas inserciones y deleciones y en la estimación del número de copias. Los reads alineados de forma aberrante, es decir, en los que los reads de un par muestran una distancia o una orientación inesperada, se analizan como indicadores potenciales de variantes estructurales. Por último, el acoplamiento de novo de reads no alineados con la referencia proporciona predicciones de variantes estructurales y de nuevas inserciones.
Adaptado a partir de Koboldt et al.17.
CNV: copy number variants (variantes variantes de número de copia); Indels: inserciones y deleciones; SNP: single nucleotide polymorphisms (polimorfismos de nucleótido único); VE: variantes estructurales.
Actualmente son tres las tecnologías de NGS mayoritariamente utilizadas por la comunidad científica7. Aunque hay otras tecnologías de secuenciación (de segunda generación), ninguna de ellas ha demostrado (hasta el momento) ser tan prominente como los instrumentos 454 GSFlex de Roche10,11, Genome Analyzer o HiSeq de Illumina12,13 y SOLiD de Life Technologies14. En los dos últimos años estos instrumentos han sufrido tantas mejoras y de forma tan rápida que ninguno de los actuales se parece al que fue lanzado comercialmente con el mismo nombre, con excepción de la química de secuenciación básica subyacente. Recientemente han aparecido versiones de equipos de NGS con características más limitadas en cuanto a su rendimiento y capacidad de secuenciación pero con mayor facilidad de manejo y enfocados a un segmento de mercado distinto. Algunos ejemplos son los equipos 454 GS Junior de Roche, MiSeq Personal Sequencer de Illumina e Ion Torrent de Life Technologies (tabla 1).
Una parte muy importante en el esquema de trabajo de un experimento de NGS es el análisis computacional (figs. 2A y B). Las ciencias informáticas han tomado una relevancia crítica en la NGS en el sentido de que sus capacidades son esenciales para manejar y analizar datos biológicos15. La NGS produce una cantidad de datos sin precedentes que un ordenador común no puede manejar5. Aunque para algunas plataformas existen herramientas de manejo de datos y análisis en un único programa, cualquier tarea no trivial a realizar con los datos requerirá al menos de una persona con conocimientos en bioinformática. En el futuro las compañías de software y los proveedores de equipos de NGS desarrollarán programas con los que no será imprescindible tener conocimientos en bioinformática para analizar datos de secuenciación masiva, aunque este hecho podría limitar al usuario a solo aprovechar las funciones predefinidas en ese hipotético software16. Además, un análisis de los datos experimentales que tenga sentido biológico depende de la incorporación de toda la información relevante que exista. Por tanto, nos parece evidente que, a día de hoy, para analizar los datos experimentales obtenidos e integrarlos con la información disponible en las numerosas bases de datos de información biológica, los conocimientos en ciencias computacionales son absolutamente necesarios.
Utilidad de la secuenciación de nueva generación para detectar variantes genéticasLa detección de variantes genéticas a partir de datos de NGS consiste en identificar diferencias en la secuencia de ADN de un individuo al compararlo con un ADN de referencia. Los resultados dependen forzosamente de la calidad del alineamiento y ensamblaje respecto a la referencia ya que las secuencias alineadas incorrectamente pueden producir falsos positivos, mientras que las secuencias no alineadas pueden ser fuente de falsos negativos. La NGS tiene el potencial de detectar cualquier tipo de variante genómica en un único experimento17, incluso puede detectar inversiones, una clase de variación cuyo estudio resulta muy complicado para la mayoría de las otras técnicas18.
Variantes de nucleótido únicoLa detección de variantes de nucleótido único (single nucleotide variants [SNV]) ha demostrado ser factible con una gran precisión cuando hay al menos una cobertura de 10-15 veces para la posición de la SNV y la tasa de error de secuenciación es razonable19,20. La mayoría de los algoritmos informáticos utilizados para detectar SNV emplean modelos bayesianos, calculando la probabilidad condicional de los nucleótidos en cada posición21–24 según, por ejemplo, el número de reads independientes que contienen la variante, la calidad en la asignación de la base y otros parámetros15,25.
Los errores en la secuenciación son más prevalentes en la NGS que con los métodos convencionales y pueden conducir a un falso positivo en la asignación de la base. En general estos errores se producen al azar, pero determinadas plataformas parecen producir específicamente cierto tipo de errores. En la plataforma Illumina, por ejemplo, los errores correlacionan con la posición en el read, acumulándose estos con mayor frecuencia hacia el final del mismo. Por el contrario, en la plataforma Roche/454 los errores no dependen de la posición en el read pero tienden a acumularse alrededor de secuencias de homopolímeros (regiones de ADN con 6-7 o más nucleótidos idénticos consecutivos).
Inserciones y delecionesLa detección de pequeñas inserciones y deleciones (indels) a partir de datos de NGS ha demostrado ser más compleja de lo que inicialmente se podía prever, sobre todo por culpa de la limitada longitud de los reads que producen la mayoría de las plataformas. Las variantes de ganancia o pérdida de una única base son especialmente proclives a ser mal alineadas con el genoma de referencia, produciendo una elevada tasa de falsos positivos. Un alineamiento de novo regional, que requiere cálculos computacionales elevados, contribuye a mejorar la detección de indels aunque los niveles de sensibilidad y especificidad no logran acercarse a los de la detección de SNV17,25,26.
Variantes de número de copia e inversiones grandesLos primeros métodos para identificar con precisión variantes estructurales han empleado datos de secuenciación de paired-reads y mate-pairs; parejas de reads que están relacionadas aunque no son adyacentes ni complementarias (para una explicación detallada, consultar la sección anterior)9,27–30. Estas aproximaciones son una extensión del trabajo seminal realizado para caracterizar y posicionar los extremos de BAC31,32 y aprovechan el hecho que los reads de tipo mate-pair y paired-end se generan a una distancia más o menos conocida en el genoma. Cuando los reads se alinean al genoma de referencia y sus «parejas» se alinean a una distancia sustancialmente diferente del tamaño esperado o con una orientación anómala son indicativos de la presencia de variantes estructurales9. Un único mate-pair no es suficiente para predecir estas variantes debido a varias razones: 1) el tamaño de inserto real solo se conoce de forma aproximada, 2) mate-pairs incorrectamente alineados pueden asemejar la apariencia de variantes estructurales, 3) una pequeña parte de todos los mate-pairs es quimérica. Para paliar todo esto se necesita un conjunto múltiple de mate-pairs agrupados en torno a la región candidata que den soporte a cada evento putativo25,29. Los métodos para detectar variantes estructurales basados en mate-pairs no pueden identificar inserciones de tamaño mayor al del inserto ni identificar los límites exactos de la alteración.
Un método alternativo para identificar deleciones y duplicaciones es a través del empleo de los valores de profundidad de cobertura de las secuencias33–35. Asumiendo que el proceso de secuenciación es uniforme, el número de reads alineados a una región sigue una distribución Poisson y se espera que sea proporcional al número de veces que esa región aparece en el genoma9. La hipótesis es que las secuencias obtenidas se distribuyen equivalentemente sobre el genoma y por tanto aquellas regiones que contradigan la hipótesis son candidatas a presentar cambios en su número de copias. Un factor de confusión en esta estrategia es el sesgo en la secuenciación que tienen las plataformas y específicamente el sesgo no lineal debido al contenido en GC12,35. Además los reads «mal» alineados en regiones que por ejemplo son ricas en repeticiones o altamente homólogas (por ejemplo las duplicaciones segmentarias) dificultan la identificación de variantes de número de copias (copy number variants [CNV]). Por tanto los métodos basados en la profundidad de cobertura son más adecuados para detectar las CNV más grandes sobre las cuales los diferentes sesgos posibles quedarían equilibrados al promediarse los valores25,33. En algunos estudios se han determinado las CNV mediante la comparación relativa de la cobertura entre dos genomas, de forma similar a los métodos de array-CGH36,37. Esta estrategia es la que se ha utilizado en algunos estudios prenatales no invasivos para detectar aneuploidías fetales con éxito (en el presente artículo estos estudios son comentados más adelante). En un estudio reciente la profundidad de cobertura se ha combinado con el empleo de mate-pairs para predecir CNV con mayor precisión38.
Un tercer método emplea splits-reads para detectar los puntos de rotura de CNV25,39. Esta estrategia se basa en la idea de que los reads que cubren un punto de rotura de una CNV no se van a alinear adecuadamente al genoma de referencia. Los reads que no pueden ser alineados se separan en dos partes de tal manera que cada parte se alinea a un locus diferente en el genoma. La distancia entre esas partes es indicativa de presencia de CNV o de inversiones. De forma similar a los mate-pairs, un único evento no es suficiente para predecir un punto de rotura y la agrupación de múltiples eventos es la que será más informativa. Para el empleo de este método es crítico el tamaño de los reads, ya que la probabilidad de que una secuencia de nucleótidos sea única en el genoma disminuye de manera drástica cuando su tamaño baja por debajo de los 25pb25.
Aplicaciones de la secuenciación de nueva generación en el diagnóstico molecularActualmente la mayoría de las aplicaciones de la secuenciación masiva están dirigidas a responder preguntas de investigación. No obstante la tecnología NGS promete ser muy relevante en la identificación de factores de susceptibilidad con finalidad preventiva, en estudios de farmacogenómica para determinar respuesta a fármacos y en la realización de pruebas genéticas para diagnóstico y evolución de las enfermedades40. En todas estas áreas la tecnología NGS está siendo evaluada en infinidad de estudios de prueba de concepto41–43.
Resecuenciación dirigidaSe considera resecuenciación la aplicación en la que se secuencia una porción del genoma conocida y los reads generados se alinean con un genoma de referencia conocido. Un ensayo dirigido a los loci de interés se puede optimizar para estudiar un único trastorno causado por mutaciones en múltiples genes. Un diseño específico de este tipo puede optimizarse para mejorar la fiabilidad en la detección de variantes. Esta estrategia tiene la ventaja de que minimiza la posibilidad de hallazgos que no estén relacionados con la indicación de la prueba inicial. No obstante, al estar identificándose nuevos genes asociados con enfermedad, un diseño fijo podría necesitar ser actualizado frecuentemente para introducir los nuevos descubrimientos. Además, para ser realmente efectiva en costes, es necesario multiplexar muestras y secuenciarlas en un mismo experimento. Cuando se trata de una enfermedad relativamente rara esta puede no ser una opción válida u ocasionar retrasos largos antes de poder realizar la prueba para establecer un diagnóstico molecular.
Resecuenciación de exomaSecuenciar el exoma (la porción codificante del genoma humano) del paciente es otra estrategia utilizada con fines diagnósticos41. A pesar de que actualmente es más cara que secuenciar un grupo reducido y específico de genes, es también mucho más barata que secuenciar un genoma completo. Una ventaja de la secuenciación del exoma es que constituye una prueba única y similar para todos los pacientes, y que no necesitaría ser actualizada cada vez que se descubriera un nuevo gen como causa de una enfermedad concreta. Esto es beneficioso para enfermedades raras ya que no es imprescindible un número mínimo de pacientes con una determinada enfermedad para reducir los costes de elaborar un ensayo específico para esa enfermedad. También hay que tener en consideración que los diagnósticos clínicos no son siempre correctos y que los fenotipos pueden variar sustancialmente, con lo que la información generada en un único experimento podría ser revisitada si fuera necesario. La secuenciación de exoma permite una aproximación sin sesgos al diagnóstico genético que podría revelar muchos casos en los que el fenotipo no se corresponde al fenotipo clínico estándar asociado a la enfermedad43,44. Una desventaja respecto a diseños dirigidos a genes específicos es que la secuenciación de exoma no permite mucha más optimización y por lo tanto en algunos casos puede resultar muy difícil realizar el diagnóstico con fiabilidad de un grupo completo de genes conocidos. Las regiones ricas en contenido de GC por ejemplo no están bien cubiertas en general, lo cual dificulta la identificación de variantes en esas regiones.
Secuenciación de genoma completoLa secuenciación de un genoma completo se implantará en el diagnóstico cuando su rendimiento, precisión y tiempo de ejecución la hagan factible. Aunque el coste económico de la secuenciación en sí misma pueda ser asumible, el verdadero reto es combinar la secuenciación del genoma humano con una interpretación eficiente y fiable45. Algunas opciones para paliar este hecho pueden ser 1) centrarse inicialmente solo en la interpretación de mutaciones conocidas, ya que el análisis de un genoma completo es muy laborioso46,47; 2) concentrarse en el análisis de genes conocidos solamente. Esta opción eludiría hallazgos de significado incierto, al mismo tiempo que se obtendría un gran rendimiento diagnóstico. No obstante esta opción no tiene en cuenta posibles diagnósticos erróneos e impediría la identificación de nuevas variantes/regiones y nuevos mecanismos causantes de enfermedad. A tenor del mayor coste, la complicación del análisis e interpretación, y de la distribución conocida de las mutaciones causantes de enfermedad, mayoritariamente localizadas en la porción codificante del genoma, muy probablemente sea aconsejable empezar con estrategias de análisis dirigidas al principio. En el momento en que no se obtenga un resultado diagnóstico con estrategias dirigidas podría estar justificado optar por continuar el análisis con una aproximación no sesgada.
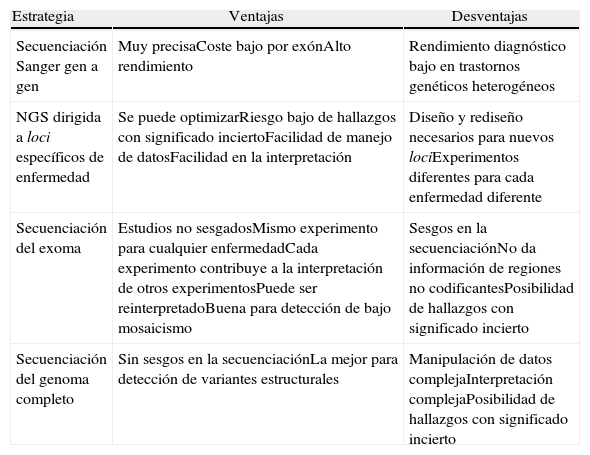
Cada una de las estrategias mencionadas exhibe ventajas específicas (tabla 2). Una estrategia dirigida parece más apropiada en enfermedades que están muy bien definidas clínicamente, para las cuales además se conoce la mayoría de los genes implicados y muestran heterogeneidad genética muy baja. La secuenciación de exoma, al ser una aproximación no sesgada, es más apropiada para enfermedades que tienen heterogeneidad fenotípica y genética mayores. Este es el método elegido en muchos casos, hasta que la secuenciación de genoma completo sea más asequible como herramienta diagnóstica y se superen las limitaciones que supone la producción de secuencias a gran escala y los desafíos bioinformáticos derivados.
Ventajas y desventajas de diferentes estrategias de secuenciación
| Estrategia | Ventajas | Desventajas |
| Secuenciación Sanger gen a gen | Muy precisaCoste bajo por exónAlto rendimiento | Rendimiento diagnóstico bajo en trastornos genéticos heterogéneos |
| NGS dirigida a loci específicos de enfermedad | Se puede optimizarRiesgo bajo de hallazgos con significado inciertoFacilidad de manejo de datosFacilidad en la interpretación | Diseño y rediseño necesarios para nuevos lociExperimentos diferentes para cada enfermedad diferente |
| Secuenciación del exoma | Estudios no sesgadosMismo experimento para cualquier enfermedadCada experimento contribuye a la interpretación de otros experimentosPuede ser reinterpretadoBuena para detección de bajo mosaicismo | Sesgos en la secuenciaciónNo da información de regiones no codificantesPosibilidad de hallazgos con significado incierto |
| Secuenciación del genoma completo | Sin sesgos en la secuenciaciónLa mejor para detección de variantes estructurales | Manipulación de datos complejaInterpretación complejaPosibilidad de hallazgos con significado incierto |
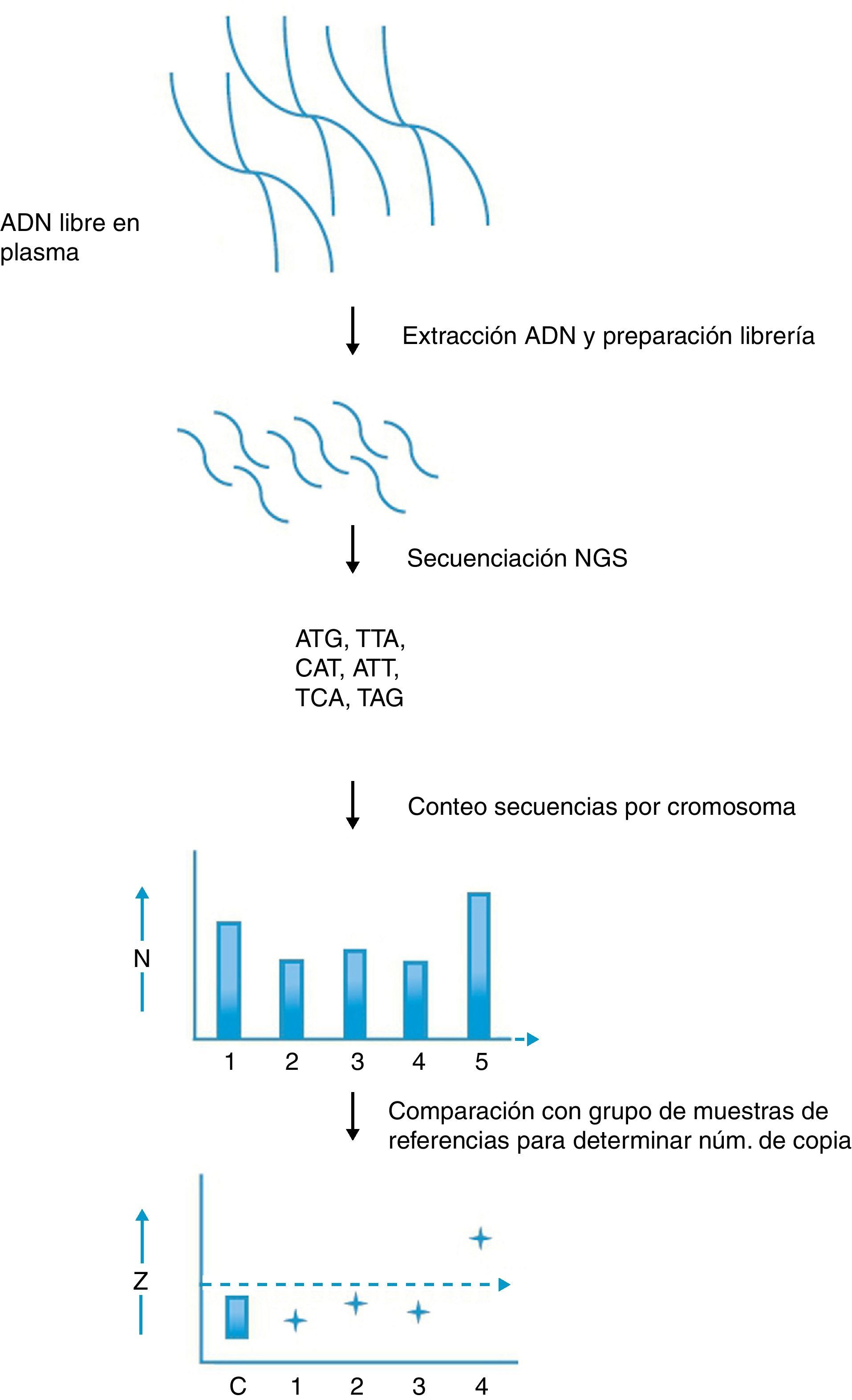
El empleo de la NGS en diagnóstico prenatal ha demostrado por el momento ser eficaz en la detección de aneuploidías a partir de ADN fetal presente en el plasma sanguíneo de la madre. La aplicación de la NGS al diagnóstico no invasivo de aneupoidías fue demostrada como prueba de concepto en dos estudios publicados en el año 200848,49. Ambos grupos demostraron que es posible detectar una trisomía del cromosoma 21 mediante datos de secuenciación masiva con gran especificidad y sensibilidad. Brevemente, el método empleado consiste en alinear los reads al genoma de referencia y realizar conteos de aquellos que se alinean en cada cromosoma para, a continuación, calcular el número de copias relativo de los cromosomas (fig. 2). En el caso de una trisomía 21, u otras aneuploidías, el número de copias relativo del cromosoma alterado debe estar significativamente sobre-representado en el conjunto de datos. Estudios posteriores han logrado optimizar los algoritmos y métodos de análisis hasta llegar a un 100% de acierto en el diagnóstico de trisomías de los cromosomas 21 y 1850. A pesar de ser estudios muy prometedores, estos trabajos están realizados en un grupo de muestras relativamente reducido. En un trabajo reciente se ha estudiado la eficacia clínica y la viabilidad práctica de la secuenciación masiva del ADN fetal de plasma materno para detectar trisomías 21 utilizando un número de muestras grande. La cohorte utilizada en este estudio fue de 753 mujeres embarazadas con indicación clínica de realización de una amniocentesis o toma de muestra de vellosidad coriónica debido a un riesgo alto de padecer trisomía 21 fetal51. La precisión diagnóstica se validó con el cariotipo utilizándose muestras de plasma maternas archivadas o recogidas prospectivamente. El método de secuenciación masiva se realizó con dos protocolos distintos para evaluar diferentes niveles de paralelización (multiplexado) de muestras: 2-plex y 8-plex. Los resultados principales indican valores de sensibilidad y especificidad del 100 y 97,9% y de 79,1 y 98,9% para la 2-plex y la 8-plex, respectivamente. Los valores predictivos positivos fueron del 96,6 y 79,1%, mientras que los negativos fueron del 100 y 98,9%. Si las muestras hubieran sido referidas para amniocentesis o muestra de vellosidad coriónica según los resultados de secuenciación masiva se habría podido evitar casi el 98% de las pruebas de diagnóstico invasivas51. En otro estudio en el que se ha explorado la utilización de la NGS para identificar trisomías 13 y 18 en una población amplia (392 embarazos) se identificó inicialmente el 36 y el 73% de trisomías 13 y 18, con unas especificidades del 92,4 y el 97,2%, respectivamente52. La detección de trisomías 13 aumentó al 100% con una especificidad del 98,9% después de utilizar como referencia un genoma sin enmascarar las repeticiones y de corregir los datos de secuencia por el contenido de GC. Los valores para la trisomía 18 cambiaron a 91,9 y 98%. Estos resultados indican que un análisis bioinformático adecuado contribuye crucialmente en el diagnóstico prenatal no invasivo de estas dos trisomías (fig. 3)52.
 permite determinar la dotación cromosómica. Adaptado de Hanh et al.54.")
Detección de aneuploidías fetales empleando secuenciación de nueva generación. En este procedimiento se aíslan los fragmentos de ADN fetal libres en el plasma materno y se produce una librería con secuencias adaptadoras especiales. Estos adaptadores permiten el consiguiente análisis múltiple. La librería se somete a secuenciación masiva para determinar la secuencia de cada fragmento. Las secuencias se alinean con el genoma de referencia y se identifica su localización cromosómica mediante métodos bioinformáticos. A continuación se cuenta el número de reads para cada cromosoma. Para el cromosoma 21 normalmente se obtienen varios miles de reads, los cuales pueden entonces compararse con los otros millones de reads distribuidos a lo largo del genoma. Si el feto está afectado con síndrome de Down el número de reads asignados al cromosoma 21 será ligeramente mayor en comparación con el número obtenido a partir de fetos normales. La comparación de estos datos con un banco de muestras de referencia y el empleo de valores prefijados de corte (Z score) permite determinar la dotación cromosómica.
Adaptado de Hanh et al.54.
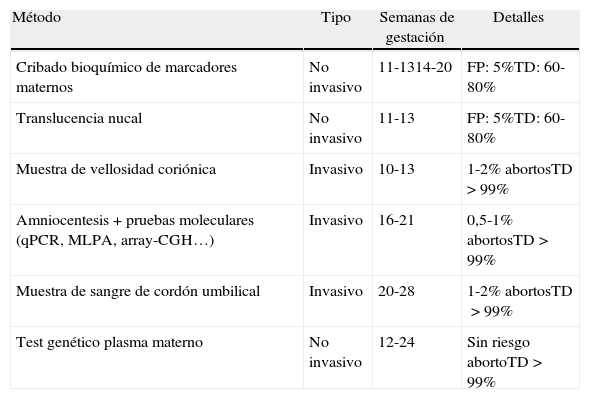
No es difícil imaginar que la detección de una aneupolidía en un diagnóstico no invasivo utilizando NGS es relativamente sencilla gracias al tamaño de la anomalía (un cromosoma entero). Sin embargo, también existen evidencias de que la NGS puede aplicarse con éxito en la detección de síndromes de microdeleción de tamaño mucho más reducido. Este es el caso de la detección de una deleción de 4,2Mb en 12p11.22 en el feto en una familia en la que el padre era portador de la alteración y estaba diagnosticado con síndrome de Asperger, además de exhibir rasgos faciales dismórficos, braquidactilia y estatura baja. La pareja ya había tenido un hijo portador de la deleción que mostraba retraso en el desarrollo y rasgos dismórficos. En el caso del embarazo descrito en el artículo, se realizó una amniocentesis y una prueba con microarrays que detectó la misma deleción en heterocigosis en el feto. El plasma de la madre se empleó entonces para completar exitosamente un estudio de prueba de concepto de uso de la NGS para detectar síndromes de microdeleción utilizando métodos de análisis similares a los utilizados para detectar aneuploidías53. No es osado pensar que esta misma aproximación puede ser válida para identificar otros síndromes conocidos que ocurren en otros loci del genoma humano. La NGS presenta por tanto ventajas considerables para ser considerada una opción razonable para detectar aneuploidías y otras alteraciones de número de copia relevantes (tabla 3).
Comparativa entre pruebas prenatales
| Método | Tipo | Semanas de gestación | Detalles |
| Cribado bioquímico de marcadores maternos | No invasivo | 11-1314-20 | FP: 5%TD: 60-80% |
| Translucencia nucal | No invasivo | 11-13 | FP: 5%TD: 60-80% |
| Muestra de vellosidad coriónica | Invasivo | 10-13 | 1-2% abortosTD >99% |
| Amniocentesis+pruebas moleculares (qPCR, MLPA, array-CGH…) | Invasivo | 16-21 | 0,5-1% abortosTD>99% |
| Muestra de sangre de cordón umbilical | Invasivo | 20-28 | 1-2% abortosTD>99% |
| Test genético plasma materno | No invasivo | 12-24 | Sin riesgo abortoTD>99% |
FP: falsos positivos; TD: tasa de detección.
Por tanto, la aplicación de la NGS ha demostrado su efectividad en estrategias no invasivas que detectan con éxito trisomías 21, 13, 18 y síndromes de microdeleción. Aunque estos adelantos técnicos son muy prometedores todavía son muy complejos y costosos en su forma actual para la mayoría de los laboratorios54. Es necesario simplificar considerablemente los procedimientos para optimizar su traslación a la clínica.
ConclusionesLa demanda creciente para disponer de técnicas de secuenciación a bajo coste ha conducido al desarrollo de tecnologías de secuenciación que producen miles de millones de secuencias de forma simultánea55. Estas tecnologías de secuenciación de alto rendimiento permiten reducir el coste de la secuenciación del ADN más allá de lo que es posible con los métodos convencionales (tabla 1). Estas nuevas tecnologías permiten una secuenciación más barata y eficiente, a pesar de obtener fragmentos (reads) de menor tamaño. Estas secuencias aisladas obtenidas requieren el empleo de potentes herramientas informáticas para su alineamiento y ensamblaje.
La irrupción de las tecnologías de secuenciación de nueva generación en la genética molecular promete superar todas las limitaciones de las estrategias actuales utilizadas para la identificación de variantes y genes asociados a enfermedad. Las características de estas tecnologías pueden contribuir sustancialmente a mejorar el proceso de diagnóstico molecular de enfermedades causadas por variantes genéticas. Las técnicas actuales de diagnóstico implican el cribado de mutaciones en loci únicos escogidos según el fenotipo clínico del paciente. Según el fenotipado o diagnóstico clínico puede ser necesario realizar pruebas en uno o en múltiples loci56. La NGS necesita menos mano de obra y menos tiempo que un protocolo estándar basado en la secuenciación convencional con el método de Sanger para estudiar trastornos heterogéneos genéticamente57,58. La corriente principal de uso de la genética en Medicina demanda que las pruebas genéticas sean capaces de identificar cualquier mutación patogénica en un período de tiempo breve. Además los resultados deben tener alta precisión y especificidad. Salvo contadas excepciones, la NGS no se ha probado de forma extensa en laboratorios de diagnóstico y parece claro que tiene que enfrentarse a varios desafíos. La tecnología NGS está en pleno desarrollo, en continua mejora y adaptación, lo cual impide una comparación favorable respecto a procedimientos diagnósticos ya establecidos, sobre todo en términos de precisión, tiempo hasta presentar el informe, reproducibilidad y costes. A pesar de que todos estos parámetros se consideran críticos para una aplicación diagnóstica, el beneficio potencial en la mejora de los rendimientos del diagnóstico supera las desventajas que puede haber actualmente. No obstante, dado el ritmo acelerado de desarrollo y mejora de la tecnología NGS en los últimos años, estos obstáculos serán probablemente salvados muy pronto.
Conflicto de interesesB.R-S y L.A son el Director de Investigación y Desarrollo y el Director Ejecutivo, respectivamente, de qGenomics.
Damos las gracias al Dr. Christian Gilissen del departamento de Genética Humana (Radboud University Nijmegen Medical Centre, Holanda) por su ayuda en la elaboración del manuscrito.