





SARS-CoV-2 (Severe Acute Respiratory Syndrome), an etiolating agent of novel COVID-19 (coronavirus 2019) pandemic, rapidly spread worldwide, creating an unprecedented public health crisis globally. NSP5, the main viral protease, is a highly conserved protein, encoded by the genome of SARS-CoV-2 and plays an important role in the viral replication cycle. In the present study, we detected a total of 33 mutations from 675 sequences submitted from India in the month of March 2020 to April 2021. Out of 33 mutations, we selected 8 frequent mutations (K236R, N142L, K90R, A7V, L75F, C22N, H246Y and I43V) for further analysis. Subsequently, protein models were constructed, revealing significant alterations in the 3-D structure of NSP5 protein when compared to the wild type protein sequence which also altered the secondary structure of NSP5 protein. Further, we identified 9 B-cell, 10 T-cell and 6 MHC-I promising epitopes using predictive tools of immunoinformatics, out of these epitopes some were non-allergenic as well as highly immunogenic. Results of our study, however, revealed that 10 B-cell epitopes reside in the mutated region of NSP5. Additionally, hydrophobicity, physiochemical properties, toxicity and stability of NSP5 protein were estimated to demonstrate the specificity of the multiepitope candidates. Taken together, variations arising as a consequence of multiple mutations may cause alterations in the structure and function of NSP5 which generate crucial insights to better understand structural aspects of SARS-CoV-2. Our study also revealed, NSP5, a main protease, can be a potentially good target for the design and development of vaccine candidate against SARS-CoV-2.
El SARS-CoV-2 (Síndrome Respiratorio Agudo Severo), un agente etiológico de la nueva pandemia de COVID-19 (coronavirus 2019), se propagó rápidamente por todo el mundo y creó una crisis de salud pública sin precedentes a nivel mundial. El NSP5, la proteasa viral principal, es una proteína altamente conservada, codificada por el genoma del SARS-CoV-2 y juega un papel importante en el ciclo de replicación viral. En el presente estudio se detectaron un total de 33 mutaciones de 675 secuencias presentadas desde la India en el mes de marzo de 2020 a abril de 2021. De 33 mutaciones, se seleccionaron 8 mutaciones frecuentes (K236R, N142L, K90R, A7V, L75F, C22N, H246Y e I43V) para su posterior análisis. Posteriormente, se construyeron modelos proteicos que revelaron alteraciones significativas en la estructura 3D de las proteínas NSP5 en comparación con la secuencia de proteínas de tipo silvestre que también alteraron la estructura secundaria de la proteína NSP5. Además, se identificaron 9 epítopos prometedores de células B, 10 de células T y 6 de MHC-I, utilizando herramientas predictivas de inmunoinformática, algunos no alergénicos y altamente inmunogénicos. Los resultados de nuestro estudio, sin embargo, revelaron que 10 epítopos de células B residen en la región mutada de NSP5. Adicionalmente, se estimó la hidrofobicidad, propiedades fisicoquímicas, toxicidad y estabilidad de la proteína NSP5 para demostrar la especificidad de los candidatos multiepítopos. En conjunto, las variaciones que surgen como consecuencia de múltiples mutaciones pueden causar alteraciones en la estructura y función del NSP5 que generan conocimientos cruciales para entender mejor los aspectos estructurales del SARS-CoV-2. Nuestro estudio también reveló que el NSP5, una proteasa principal, puede ser un blanco potencialmente bueno para el diseño y desarrollo de la vacuna candidata contra el SARS-CoV-2.
The rapid emergence of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), responsible for causing the ongoing pandemic of novel coronavirus disease 2019 (COVID-19), induces moderate to severe respiratory distress (such as cough, cold, dyspnea) in humans around the world.1 The novel COVID-19 has been reported from the wildlife market in Wuhan city of Hubei province (China), in late December 2019.2 SARS-CoV-2 has now affected 218 countries, posing devastating public health threat across the globe. As of May 21, 2021, almost after 18 months of the outbreak of this pandemic worldwide over 107,838,255 confirmed cases of COVID-19 have been reported to WHO including 2,373,398 casualties (WHO COVID-19 Dashboard).3 As many as 60 different vaccines against coronavirus have reached various stages of clinical development and many of them have been approved for immunization purposes nowadays. Vaccinating vulnerable population to achieve herd immunity against SARS-CoV-2 infection is of great importance, however, due to the emergence of new variants of this virus it is very difficult to assess how long the available vaccine will remain durable and effective.4,5
Coronavirus (CoVs) is an enveloped, single-stranded, positive sense RNA virus of ~30 kb length .6 The genome of SARS-CoV-2 encodes four types of structural (spike S, envelope E, membrane M and nucleocapsid N) and various conserved non-structural proteins ranging from NSP1to NSP16 including nine accessory proteins.7,8 ORF1ab encodes non-structural proteins of SARS-CoV-2 which is crucial for the viral life cycle and pathogenesis. NSP5 (main viral protease, Mpro) has been found synonymous with 3C-like protease (3CLpro), that mediates cleavage at 11 different sites of polyproteins to generate other non-structural proteins and also plays significant role in the viral replication cycle.9,10 Due to its essential and conserved role in viral development, NSP5 is considered as promising antiviral therapeutic target against SARS-CoV-2 infections. NSP5 of coronavirus is a ~30 KDa protein, possessing structurally conserved three domain cysteine protease and acts as a main protease for proteolytic processing of viral replicase polyproteins such as pp1a and pp1b.11–14 Interestingly, the yields of NSPs proteins gets affected by the inhibition of the NSP5-mediated cleavage and hence, the viral replication can also be prevented. Due to this reason, since the advent of this pandemic, several studies have been performed to identify various compounds, capable to antagonize the activity of NSP5 and also help in better understanding of the molecular mechanism behind the inhibition.15,16
The genome of SARS-CoV-2 is rapidly evolving by acquiring multiple mutations. As it is quite evident from numerous previous studies, NSP5 of coronavirus plays crucial role in the viral infection and pathogenesis. Present in silico study was, therefore, carried out to detect and characterize mutations of NSP5 of SARS-CoV-2. We identified a total of 33 mutations from 675 sequences submitted from India in the month of March 2020 to April 2021 and compared with the first reported sequence from Wuhan, as a reference sequence. Subsequently, the impact of mutation on the secondary structure and protein dynamics was observed that help in designing therapeutics and/or vaccine to curb SARS-CoV-2 infections.
In addition to this, NSP5 of SARS-CoV-2 was explored to determine the potent antigenic epitopes of B-cell and T-cell with their MHC alleles to predict multiepitope vaccine (MEV) construct. Owing to their specificity, stability, less time-consuming and cost-effective properties as well as the ability to induce significant humoral and cellular immune responses, MEVs are found to be advantageous over single epitope or conventional vaccine development approach.17 Further, several predictive tools of immunoinformatics were utilized to validate the non-allergenic, non-toxic, antigenicity, toxicity, structural stability/flexibility and physiochemical properties and hydrophobicity of the designed multi-epitopes vaccine candidate.
Materials and methodsData miningThe full length protein sequence of ORF1ab polyprotein, 7096 amino acid long which encodes for non-structural proteins in SARS-CoV-2 were retrieved from NCBI virus database. NCBI virus database keeps a deposit of all SARS-CoV-2 sequences submitted from different parts of the world. As on April 29, 2021, 675 full length ORF1ab amino acid sequences were submitted from India which was used in this study. The first reported ORF1ab protein sequence with Accession number YP_009724389 was also downloaded to be used as a reference or wild type sequence in this study. From the full length ORF1ab polyprotein sequence, the sequence of NSP5 (SARS-CoV-2 protease) was procured being 306 amino acid long.
Identification of protease mutants from IndiaTo detect the variations in the protease protein amino acid sequences, the NSP5 protein sequences from India were aligned with the first reported SARS-CoV-2 sequence from Wuhan. To align these polypeptides, Clustal Omega online platform18 was used which creates 1000 of alignments based on HMM profile seeded guide trees. These alignments were viewed on Jalview to detect the variations occurring in the protease protein with reference to Wuhan type protease sequence. The non-synonymous amino acid variants were analyzed using Protein Variation Effect Analyzer known as PROVEAN v1.1.3 with cutoff predicted score of −2.50 to detect the effect of mutation on the NSP5 protein.19 PROVEAN predicts the effect of amino acid substitution on the overall function of aa protein. A score namely delta alignment is calculated which are the PROVEAN scores of the substituted protein. The threshold limit for this score being −2.5 below or equal to which the mutation is deleterious and above this threshold limit the variation has neutral effect.
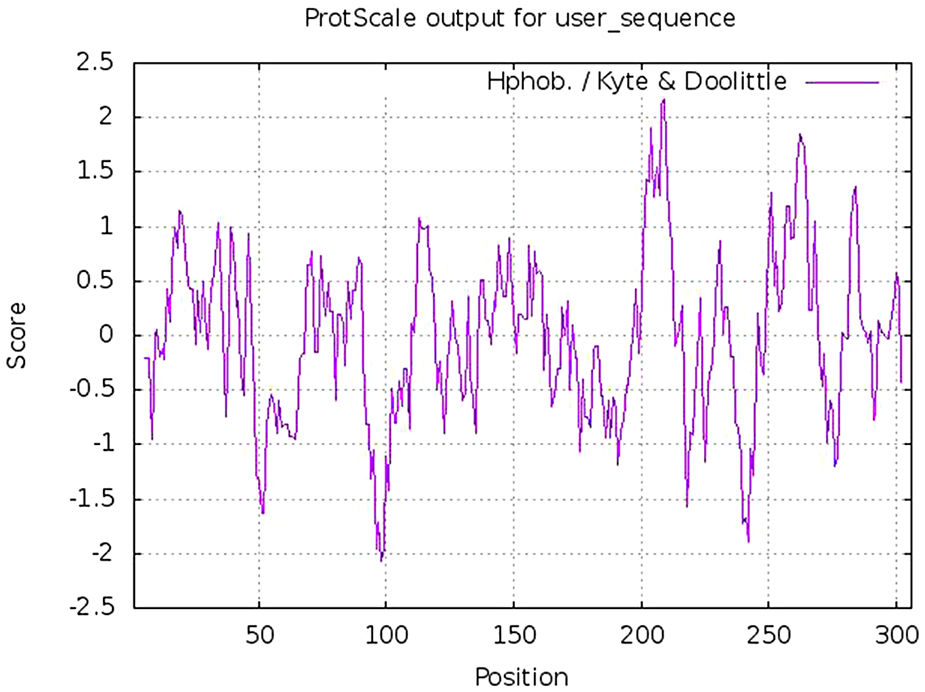
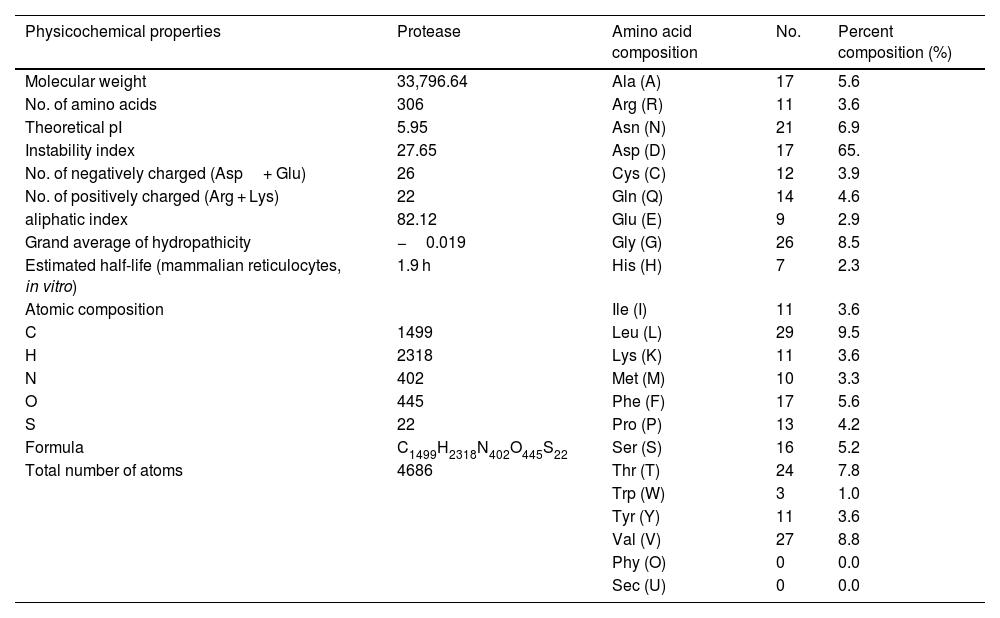
Calculation of physicochemical properties and hydropathy index of protease proteinPhysicochemical properties of any protein includes its molecular weight, aliphatic index, composition of different amino acids including positively and negatively charged, atomic composition, estimated half life, instability index, hydrophobicity (GRAVY score) and other parameters. These parameters were calculated using Protparam tool of Expasy online platform. The hydropathy plot was prepared using Protscale tool, an expasy program.20
Secondary structure predictionThe secondary structure of the NSP5 protein was predicted using CFSSP (Chou and Fasman Secondary Structure Prediction) online software.21 The analysis was done for both wild type and mutated protein sequences to study the alteration in the secondary structure of the protein such as changes in helix, turn and sheet formation due to mutation.
NSP5 protein dynamics studyPhyre2 online modeling platform was used to build the models of wild type and mutated NSP5 proteins.22 Dynamut software was applied to detect the impact of mutation on the structure flexibility and dynamicity of NSP5 protein.23 Dynamut computes information on the stability, NMA analysis, flexibility, rigidness, conformation of mutated as well as wild type protein. Several parameters were calculated like flexibility analysis, vibrational entropy, atomic and deformation energies using first 10 non-trivial modes of the structure. To check whether upon variation intramolecular interactions can change, Dynamut was used to predict the effect of mutation on intramolecular interactions.
Identification of lineal B-cell epitopesIEDB was used to predict the lineal B-cell epitopes in the NSP5 protein of SARS-CoV-2.24 IEDB webserver constructs epitopes based on estimation of parameters like flexibility, accessibility, hydrophilicity, turns, polarity and antigenic propensity of the protein using amino acid scales and HMMs.
MHC class I allele identificationThe T-cell epitope binding alongwith the detection of MHC allele showing highest affinity for the T-cell epitope was predicted using IEDB Tepitool server.24 This platform provides information on the binding of HLA allele with both type I and type II MHC molecules.
Antigenicity and allergenicity evaluationTo identify the antigenicity of the NSP5 protein, Vaxijen v2.0 server which predicts antigens according to the auto cross-covariance (ACC) transformation of the protein sequences was used.25 The prediction of vaccine allergenicity was done using AllerTOP server, which evaluates protein allergenicity on auto cross variance (ACC method) that explains residues hydrophobicity, size, flexibility and other parameters.26
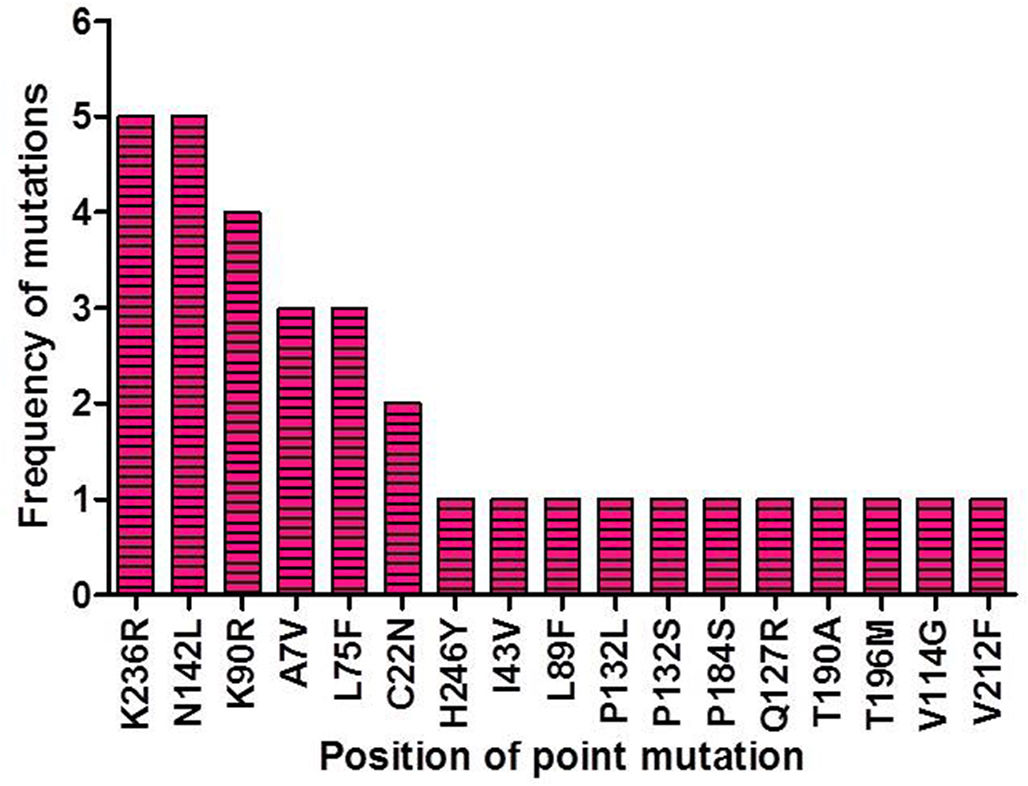
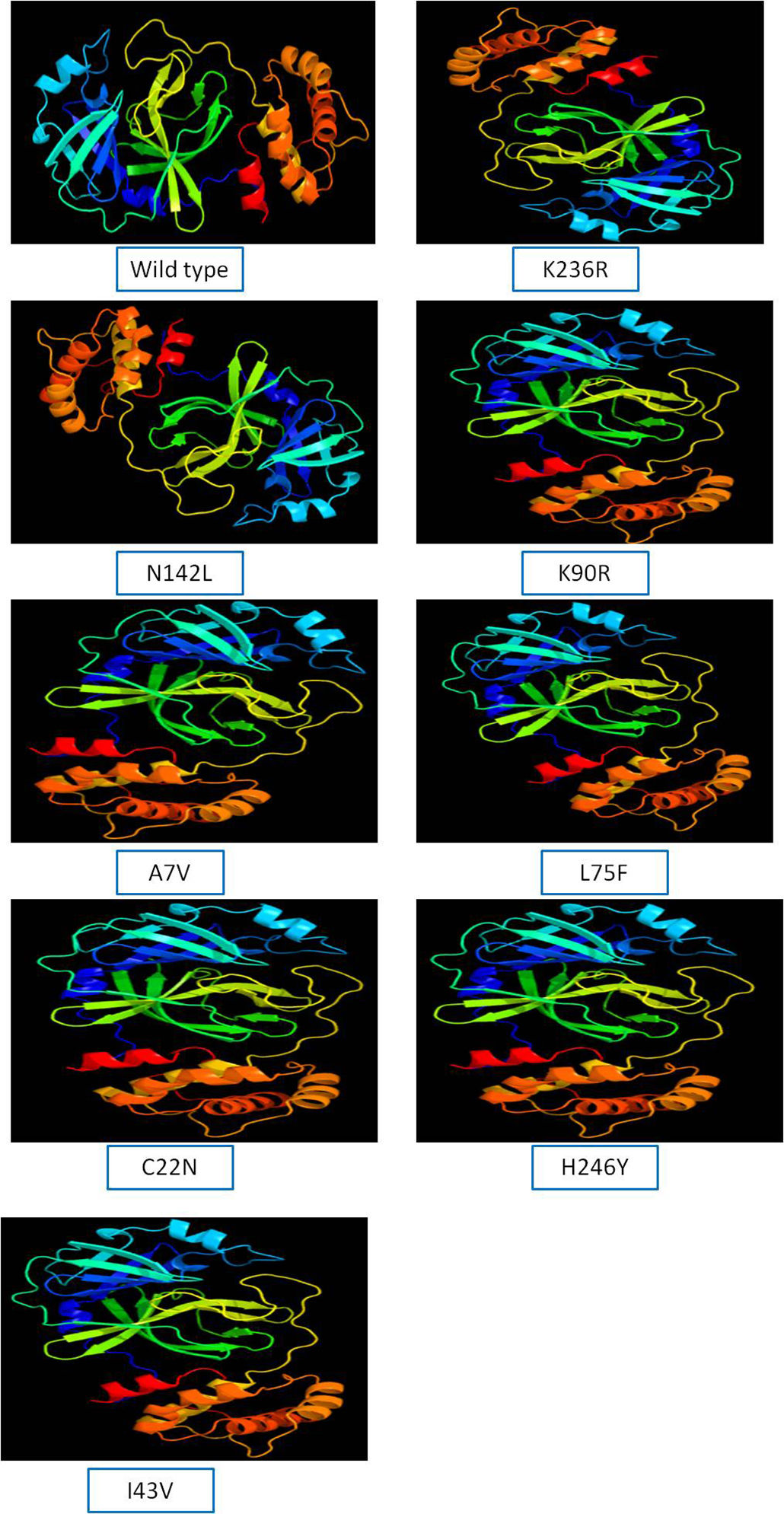
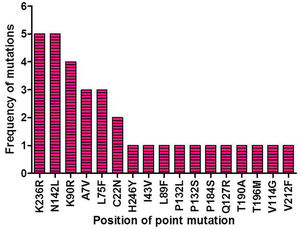
ResultsIdentification of mutation in protease of SARS-CoV-2 and detection of non synonymous mutantsAltogether 675 full length sequences of ORF1ab were submitted from India from March 2020 to April 2021. These 675 sequences were downloaded alongwith a reference sequence of Wuhan type virus from NCBI virus database (Supplementary table 1). The multiple sequence alignment was performed for all these ORF1ab sequences with reference to Wuhan type virus and the alignment file was viewed using Jalview. Those mutations which occurred in NSP5 were recorded and used for further analysis. A total of 33 point mutations were detected in this 306 amino acid long NSP5 protein of Indian isolates (Supplementary table 2). Amongst these point mutations, K236R, N142L, K90R, A7V, L75F, C22N, H246Y and I43V were the most frequently occurring mutations and hence used for further characterization in this study (Supplementary Fig. 1).
The three non-synonymous amino acid substitutions (N142, L75F and C22N) amongst the eight showed deleterious impact on the structure and function of NSP5 protein. All other five mutants showed neutral impact on the protein at −2.5 cutoff values of PROVEAN score (Supplementary table 3).
Estimation of physicochemical properties and hydropathy index of SARS-CoV-2 NSP5 proteinThe physicochemical properties of SARS-CoV-2 protease protein were estimated using Protparam (ExPasy). The analysis revealed that the NSP5 protein is 306 amino acids in length with a molecular weight of 33,796.64 Da, instability index 27.65, aliphatic index 82.12 and GRAVY score of −0.019 (Table 1). The hydropathy plot showed C-terminal amino acid to be more hydrophobic as compared to the N-terminal end of NSP5 protein (Fig. 1).
Physicochemical properties of NSP5 protein (wild type).
| Physicochemical properties | Protease | Amino acid composition | No. | Percent composition (%) |
|---|---|---|---|---|
| Molecular weight | 33,796.64 | Ala (A) | 17 | 5.6 |
| No. of amino acids | 306 | Arg (R) | 11 | 3.6 |
| Theoretical pI | 5.95 | Asn (N) | 21 | 6.9 |
| Instability index | 27.65 | Asp (D) | 17 | 65. |
| No. of negatively charged (Asp+ Glu) | 26 | Cys (C) | 12 | 3.9 |
| No. of positively charged (Arg + Lys) | 22 | Gln (Q) | 14 | 4.6 |
| aliphatic index | 82.12 | Glu (E) | 9 | 2.9 |
| Grand average of hydropathicity | −0.019 | Gly (G) | 26 | 8.5 |
| Estimated half-life (mammalian reticulocytes, in vitro) | 1.9 h | His (H) | 7 | 2.3 |
| Atomic composition | Ile (I) | 11 | 3.6 | |
| C | 1499 | Leu (L) | 29 | 9.5 |
| H | 2318 | Lys (K) | 11 | 3.6 |
| N | 402 | Met (M) | 10 | 3.3 |
| O | 445 | Phe (F) | 17 | 5.6 |
| S | 22 | Pro (P) | 13 | 4.2 |
| Formula | C1499H2318N402O445S22 | Ser (S) | 16 | 5.2 |
| Total number of atoms | 4686 | Thr (T) | 24 | 7.8 |
| Trp (W) | 3 | 1.0 | ||
| Tyr (Y) | 11 | 3.6 | ||
| Val (V) | 27 | 8.8 | ||
| Phy (O) | 0 | 0.0 | ||
| Sec (U) | 0 | 0.0 |

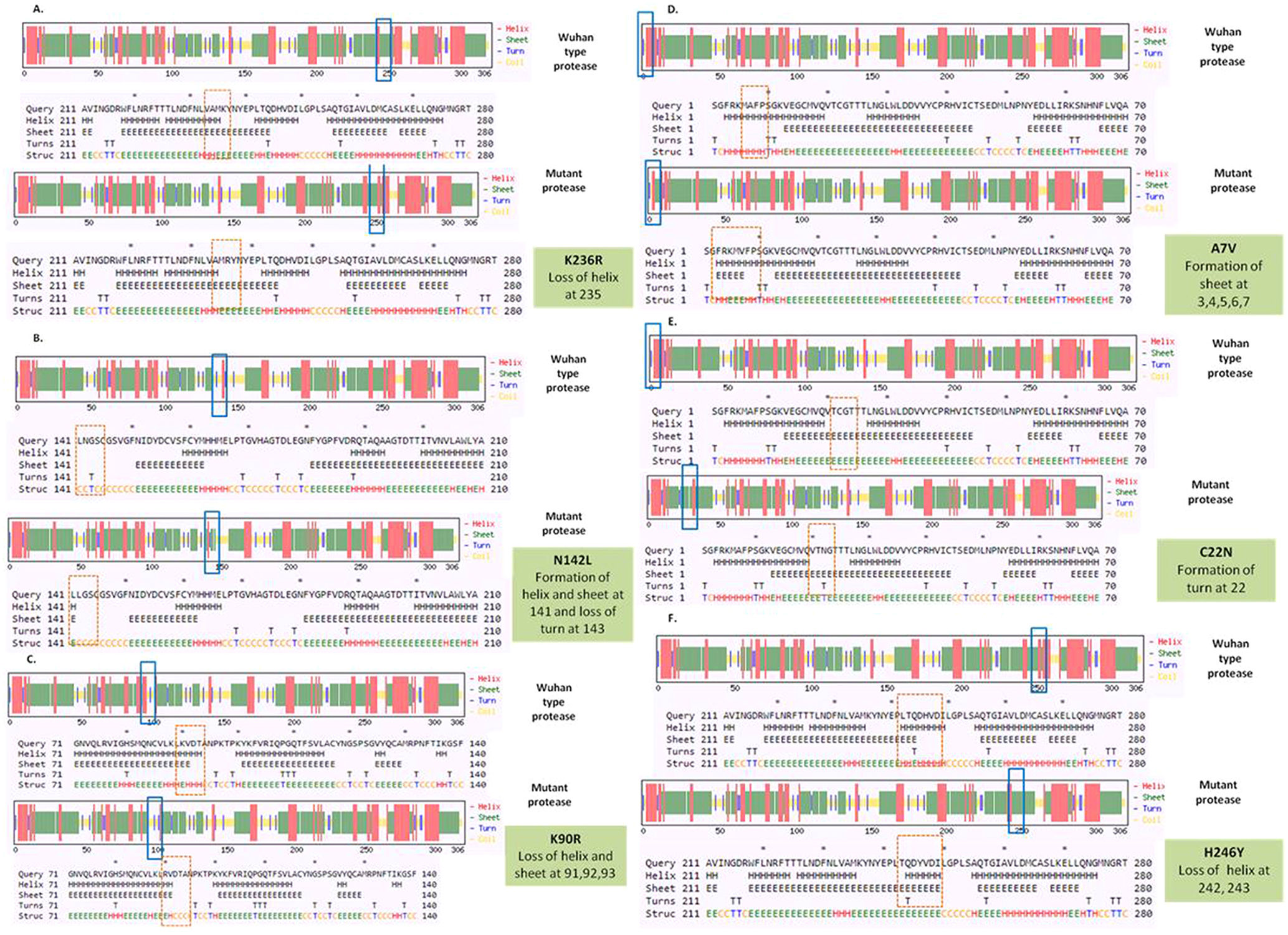
To detect the alteration in formation and loss of alpha helix, beta sheet and turns upon mutation in NSP5 protein secondary structure prediction was done using CFSSP online program with respect to wild type protein. The mutations K236R, N142L, K90R, A7V, C22N and H246Y showed significant secondary structural changes (Fig. 2a) and hence their effect was studied. The point mutation at position 236, where lysine is replaced by arginine in the NSP5 protein resulted in loss of helix structure at positions 235. Our analysis showed that the mutation at 142, where asparagine is replaced by leucine resulted in formation of helix and sheet at position 141 and loss of turn at 143. Asparagine being a polar uncharged amino acid favors formation of turn, whereas leucine being a non polar amino acid forms helix. Further, the substitution of lysine by arginine at position 90 resulted in loss of helix and sheet at points 91, 92 and 93. The A7V mutant resulted in formation of sheet at positions 3, 4, 5, 6 and 7 as valine has larger non-polar group compared to alanine and hence more tendency to form sheets. C22N mutant showed formation of turn at point 22, as asparagines favors turn formation. The substitution of histidine by tyrosine at 246 position resulted in loss of helix at 242 and 243 positions. Tyrosine being an aromatic amino acid has more tendencies to form sheets rather than helix. Overall, the secondary structure analysis depicts significant changes in the formation and loss of helix, sheet and turn that can bring huge impact on NSP5 protein and hence leading to the SARS-CoV-2 multiplication and infection.
 Secondary structure prediction of NSP5 protein. Effect of mutation at different sites on the secondary structure of protease protein (A–H). The first secondary structure in each (A–F) represents the Wuhan type sequence while the second represents the mutated one. The mutation location and respective secondary structures are marked with boxes. (b) Mutational effect on structural dynamics of protease protein. Blue represents rigidification, whereas red represents gain in flexibility upon mutation. (c) Effect of point mutation on interatomic interactions of NSP5 protein. Interatomic interactions were altered by mutations at different locations. Wild type amino acid residues are colored in light green and represented as stick with the surrounding residues where any interactions exist. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)")
 Secondary structure prediction of NSP5 protein. Effect of mutation at different sites on the secondary structure of protease protein (A–H). The first secondary structure in each (A–F) represents the Wuhan type sequence while the second represents the mutated one. The mutation location and respective secondary structures are marked with boxes. (b) Mutational effect on structural dynamics of protease protein. Blue represents rigidification, whereas red represents gain in flexibility upon mutation. (c) Effect of point mutation on interatomic interactions of NSP5 protein. Interatomic interactions were altered by mutations at different locations. Wild type amino acid residues are colored in light green and represented as stick with the surrounding residues where any interactions exist. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)")
 Secondary structure prediction of NSP5 protein. Effect of mutation at different sites on the secondary structure of protease protein (A–H). The first secondary structure in each (A–F) represents the Wuhan type sequence while the second represents the mutated one. The mutation location and respective secondary structures are marked with boxes. (b) Mutational effect on structural dynamics of protease protein. Blue represents rigidification, whereas red represents gain in flexibility upon mutation. (c) Effect of point mutation on interatomic interactions of NSP5 protein. Interatomic interactions were altered by mutations at different locations. Wild type amino acid residues are colored in light green and represented as stick with the surrounding residues where any interactions exist. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)")
(a) Secondary structure prediction of NSP5 protein. Effect of mutation at different sites on the secondary structure of protease protein (A–H). The first secondary structure in each (A–F) represents the Wuhan type sequence while the second represents the mutated one. The mutation location and respective secondary structures are marked with boxes. (b) Mutational effect on structural dynamics of protease protein. Blue represents rigidification, whereas red represents gain in flexibility upon mutation. (c) Effect of point mutation on interatomic interactions of NSP5 protein. Interatomic interactions were altered by mutations at different locations. Wild type amino acid residues are colored in light green and represented as stick with the surrounding residues where any interactions exist. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
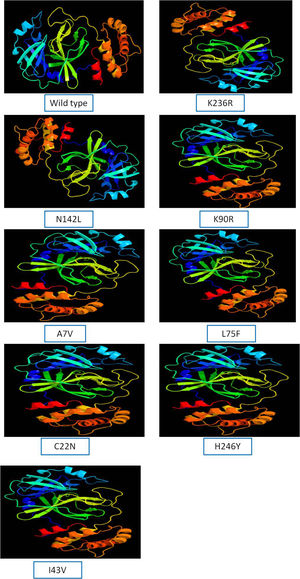
The 3D model of NSP5 protein was built using Phyre2 online modeling software, which performs modeling on the basis of template search. The template being used for protease protein was d2duca1 with 100% similarity coverage. The models of both wild type and mutated NSP5 protein sequences are shown in Supplementary Fig. 2.
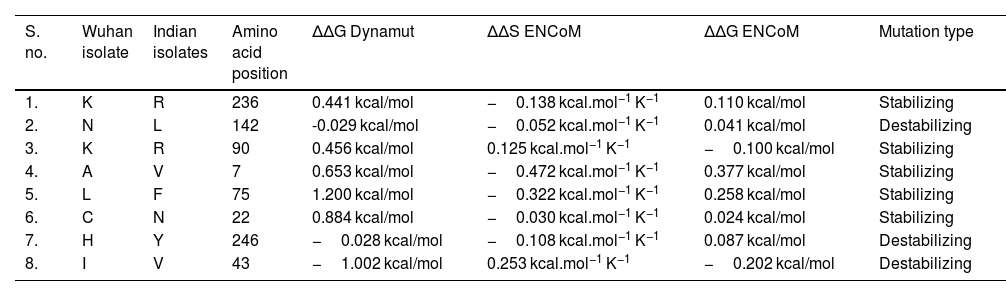
NSP5 protein flexibility and stability change upon mutationThe impact of mutation on the dynamics of protease protein was estimated using Dynamut software.23 Dynamut software estimates the flexibility or steadiness of a protein upon mutation as compared to the wild type as calculated by ENCoM, DUET, mCSM and others. Negative value of ΔΔG denotes destabilization of protein upon mutation, whereas a positive value signifies stabilization. The free energy difference, ∆∆G between the wild and mutated protein sequences was calculated using Dynamut and the values showed a stabilizing mutation in five mutants of NSP5 protein as indicated by their ∆∆G values (Table 2). The mutants N142L, H246Y and I43V were destabilizing for NSP5 protein with -∆∆G values. The most stable mutant amongst all was L75F showing highest positive value of ∆∆G (1.200 kcal/mol), followed by C22N (0.884 kcal/mol) and A7V (0.653 kcal/mol) as shown in Table 4. The highest negative value of ∆∆G was shown by I43V (−1.002 kcal/mol) followed by N142L (−0.029 kcal/mol) and H246Y (−0.028 kcal/mol). The vibrational entropy change (ΔΔSVib ENCoM) provides information on the configurational entropy of the proteins with single minima of the energy landscape. The ΔΔSVib ENCoM was calculated for the mutant and wild type protease protein to calculate the vibrational entropy energy change between wild type and mutant. The ΔΔSVib ENCoM calculated for all the protease mutants revealed a negative value signifying the rigidification of protein structure upon mutation except for I43V and K90R mutant which have positive values of ΔΔSVib ENCoM, signifying gain of flexibility upon mutation in NSP5 protein. The visual representation of flexibility analysis depicted similar results, of gain in rigidification upon mutation shown by blue region in all the NSP5 mutants except for I43V and K90R mutants, shown by red color region in Fig. 2c.
Effect of mutation on the structural dynamics of protease protein as shown by ΔΔS ENCoM and ΔΔG values.
| S. no. | Wuhan isolate | Indian isolates | Amino acid position | ΔΔG Dynamut | ΔΔS ENCoM | ΔΔG ENCoM | Mutation type |
|---|---|---|---|---|---|---|---|
| 1. | K | R | 236 | 0.441 kcal/mol | −0.138 kcal.mol−1 K−1 | 0.110 kcal/mol | Stabilizing |
| 2. | N | L | 142 | -0.029 kcal/mol | −0.052 kcal.mol−1 K−1 | 0.041 kcal/mol | Destabilizing |
| 3. | K | R | 90 | 0.456 kcal/mol | 0.125 kcal.mol−1 K−1 | −0.100 kcal/mol | Stabilizing |
| 4. | A | V | 7 | 0.653 kcal/mol | −0.472 kcal.mol−1 K−1 | 0.377 kcal/mol | Stabilizing |
| 5. | L | F | 75 | 1.200 kcal/mol | −0.322 kcal.mol−1 K−1 | 0.258 kcal/mol | Stabilizing |
| 6. | C | N | 22 | 0.884 kcal/mol | −0.030 kcal.mol−1 K−1 | 0.024 kcal/mol | Stabilizing |
| 7. | H | Y | 246 | −0.028 kcal/mol | −0.108 kcal.mol−1 K−1 | 0.087 kcal/mol | Destabilizing |
| 8. | I | V | 43 | −1.002 kcal/mol | 0.253 kcal.mol−1 K−1 | −0.202 kcal/mol | Destabilizing |
Further, the findings of our study dealt with the detection of variation in intramolecular interactions of NSP5 protein with its neighboring molecules upon mutation. All the NSP5 mutants studied here showed significant changes in intramolecular interactions that occurred in NSP5 proteins upon mutation (Fig. 2d). The mutation caused significant alterations in the interactions like hydrogen bonds, ionic interactions, hydrophobic interactions and other metal complex interactions. The substitution in side chain of the amino acids changes due to mutation hence disrupting neighboring interactions. This study predicts that the mutation in leucine, asparagines, lysine, cysteine, alanine residues causes significant alterations in the intramolecular interactions with the neighboring molecules (Fig. 2d). From these results, it can be concluded that the NSP5 protein mutation not only changes the overall dynamics of the protein but can also interrupts its intramolecular interaction.
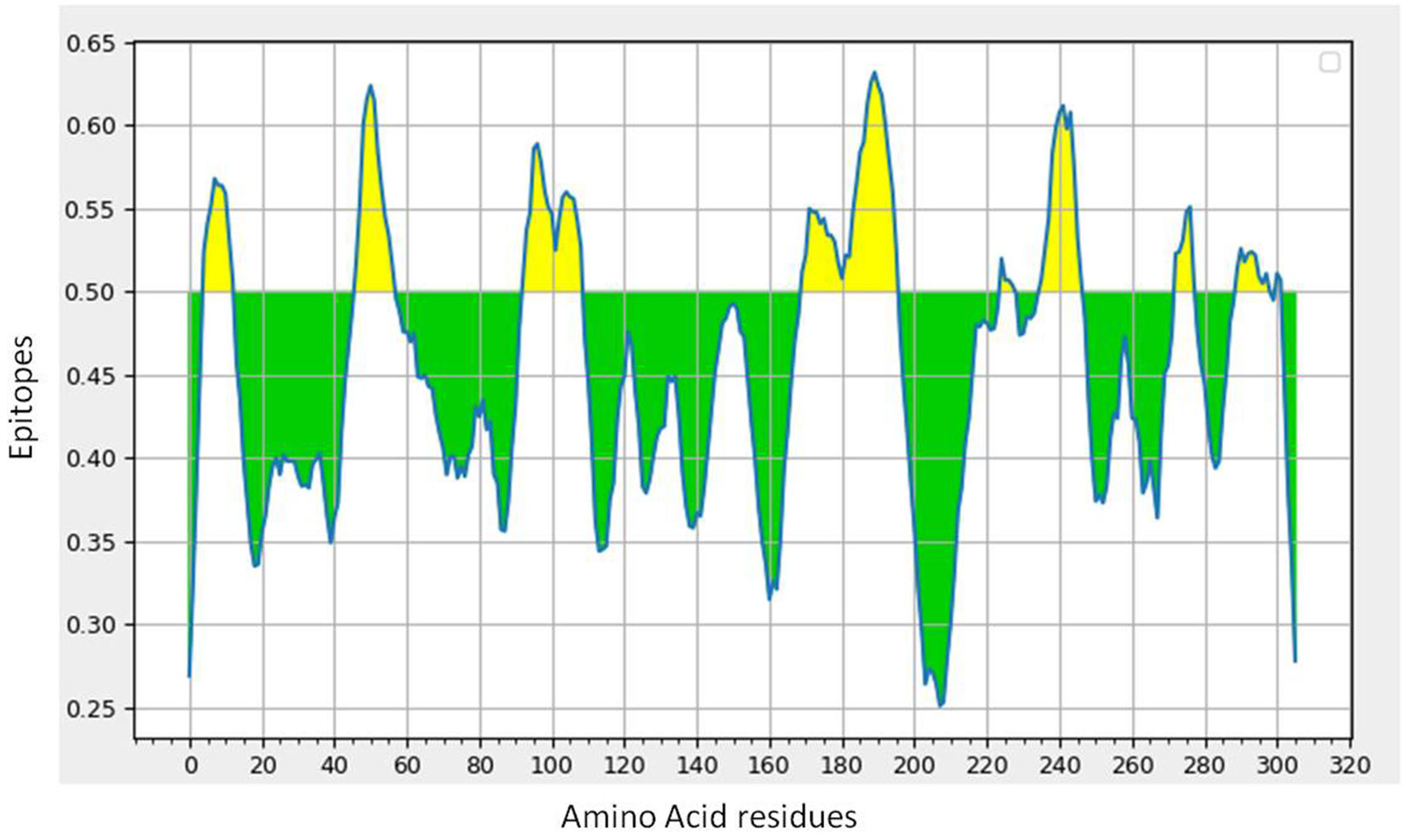
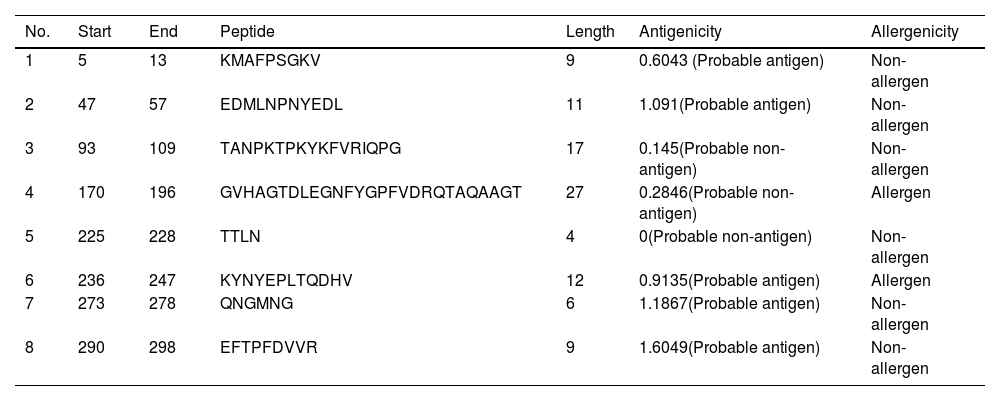
B-cell epitope prediction with its antigenicity and allergenicityLineal B-cell epitopes were predicted for NSP5 protein using NSP5 protein sequence as query and threshold value of 0.5 was selected. A total of eight B-cell epitopes predicted for this protein above the threshold value which are shown in Table 3 (Fig. 3). Out of these nine epitopes, the epitopes KMAFPSGKV, EDMLNPNYEDL, QNGMNG and EFTPFDVVR were highly antigenic as well as non-allergenic, whereas some epitopes were immunogenic but allergenic. These five predicted epitopes can be a good candidate in vaccine production against SARS-CoV-2. In our analysis, 9 mutations out of 33 were found in the epitopic region of protease protein. These mutations not only change its epitopic region rather changes its overall antigenicity and therefore can help in host evasion.
List of lineal B-cell epitopes for NSP5 protein with their sequence, length, site, antigenicity and probable allergenicity.
| No. | Start | End | Peptide | Length | Antigenicity | Allergenicity |
|---|---|---|---|---|---|---|
| 1 | 5 | 13 | KMAFPSGKV | 9 | 0.6043 (Probable antigen) | Non-allergen |
| 2 | 47 | 57 | EDMLNPNYEDL | 11 | 1.091(Probable antigen) | Non-allergen |
| 3 | 93 | 109 | TANPKTPKYKFVRIQPG | 17 | 0.145(Probable non-antigen) | Non-allergen |
| 4 | 170 | 196 | GVHAGTDLEGNFYGPFVDRQTAQAAGT | 27 | 0.2846(Probable non-antigen) | Allergen |
| 5 | 225 | 228 | TTLN | 4 | 0(Probable non-antigen) | Non-allergen |
| 6 | 236 | 247 | KYNYEPLTQDHV | 12 | 0.9135(Probable antigen) | Allergen |
| 7 | 273 | 278 | QNGMNG | 6 | 1.1867(Probable antigen) | Non-allergen |
| 8 | 290 | 298 | EFTPFDVVR | 9 | 1.6049(Probable antigen) | Non-allergen |
 B-cell epitope prediction of NSP5 protein. The threshold cutoff is 0.5 above which the residues are epitopes. (b) The results of MHC cluster analysis. (A) Heat map of MHC class I cluster, (B) tree map of MHC class I cluster. (c) The results of MHC cluster analysis. (A) Heat map of MHC class II cluster, (B) tree map of MHC class II cluster.")
 B-cell epitope prediction of NSP5 protein. The threshold cutoff is 0.5 above which the residues are epitopes. (b) The results of MHC cluster analysis. (A) Heat map of MHC class I cluster, (B) tree map of MHC class I cluster. (c) The results of MHC cluster analysis. (A) Heat map of MHC class II cluster, (B) tree map of MHC class II cluster.")
 B-cell epitope prediction of NSP5 protein. The threshold cutoff is 0.5 above which the residues are epitopes. (b) The results of MHC cluster analysis. (A) Heat map of MHC class I cluster, (B) tree map of MHC class I cluster. (c) The results of MHC cluster analysis. (A) Heat map of MHC class II cluster, (B) tree map of MHC class II cluster.")
(a) B-cell epitope prediction of NSP5 protein. The threshold cutoff is 0.5 above which the residues are epitopes. (b) The results of MHC cluster analysis. (A) Heat map of MHC class I cluster, (B) tree map of MHC class I cluster. (c) The results of MHC cluster analysis. (A) Heat map of MHC class II cluster, (B) tree map of MHC class II cluster.
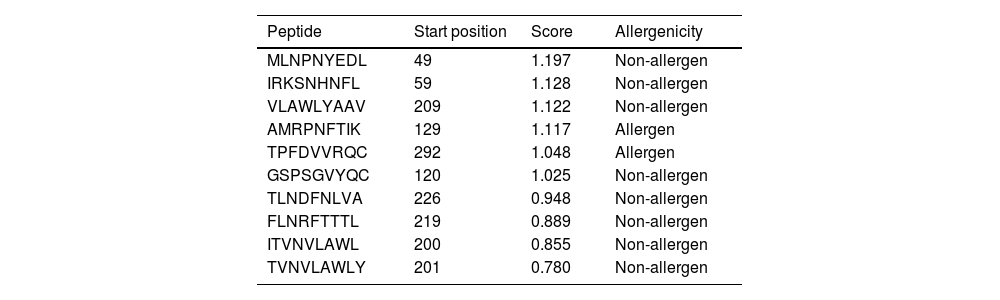
Altogether 9 T-cell binding epitopes were predicted for NSP5 protein showing different allele binding affinity. The sequence of these epitopes along with its position is shown in Table 4. Out of these nine T-cell epitopes only two were allergenic and others were immunogenic as well as non-allergenic. The MHC class I immunogenicity of the NSP5 molecules is shown in Table 5. A total of six peptides were predicted with a potential of MHC class I immunogens. These epitopes can induce immunogenicity and hence increase cytokine production in cells to combat the infection.
T-cell epitope prediction of SARS- CoV-2 protease and its allergenicity.
| Peptide | Start position | Score | Allergenicity |
|---|---|---|---|
| MLNPNYEDL | 49 | 1.197 | Non-allergen |
| IRKSNHNFL | 59 | 1.128 | Non-allergen |
| VLAWLYAAV | 209 | 1.122 | Non-allergen |
| AMRPNFTIK | 129 | 1.117 | Allergen |
| TPFDVVRQC | 292 | 1.048 | Allergen |
| GSPSGVYQC | 120 | 1.025 | Non-allergen |
| TLNDFNLVA | 226 | 0.948 | Non-allergen |
| FLNRFTTTL | 219 | 0.889 | Non-allergen |
| ITVNVLAWL | 200 | 0.855 | Non-allergen |
| TVNVLAWLY | 201 | 0.780 | Non-allergen |
Showing class I immunogenicity of NSP5 protein of SARS-CoV-2.
| Peptide | Length | Score |
|---|---|---|
| FYGPFVDRQTAQAAGTDTTITVNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYE | 60 | 1.51334 |
| SGVTFQ | 6 | 0.16646 |
| PLTQDHVDILGPLSAQTGIAVLDMCASLKELLQNGMNGRTILGSALLEDEFTPFDVVRQC | 60 | 0.1167 |
| SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTSEDMLNPNYEDLLIR | 60 | −0.01126 |
| SPSGVYQCAMRPNFTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGN | 60 | −0.11804 |
| KSNHNFLVQAGNVQLRVIGHSMQNCVLKLKVDTANPKTPKYKFVRIQPGQTFSVLACYNG | 60 | −0.9389 |
The cluster analysis of MHC class I allele is shown in Fig. 3c while that of class II allele is shown in Fig. 3d, where the red zone denotes strong interaction of the HLA allele with the epitopes of NSP5 protein, whereas yellow depicts weak interaction. We analyzed the binding ability of all the alleles with the protease epitopes.
Assessment of antigenicity and allergenicityVaxiJen v2.0 server was used to predict the antigenicity of the protease protein. The property of antigenicity depends on the ability of the vaccine to bind to the B-cell and T-cell receptors and increase the immune response in the cell. This analysis indicates that the NSP5 protein sequence is antigenic with potent antigenicity at a threshold of 0.4%. A good immunogen should not show allergic response in the host cell. The allergenicity of B-cell epitopes of the NSP5 protein was predicted using Allertop tool as many B-cell and T-cell epitopes were non-allergenic and hence can be a candidate protein for vaccine development.
DiscussionCoronavirus poses an unprecedented threat for human health globally. Considering its contagiousity, World Health Organization on March 11, 2020 has declared public health emergency internationally (WHO 2020). SARS-CoV-2 is a member of RNA viruses and has remarkable capacity to mutate their genome in a very short period of time.27 Notably, majority of viral mutation shows harmful effects. Moreover, a mutation is essential for viral evolution and adaptability, these traits are considered as the key determinants for viruses to survive in the dynamic environment of host and also enabling them to evade the pre-existing immunity of host and most often acquire drug resistance. SARS-CoV-2 infections emerged from Wuhan, China, soon began to spread globally. Rapid transmission of coronavirus infection depends on various factors such as polymerase fidelity, different geographical areas and population density, as well as poor health care system, climatic and environmental variations.28 Mutational analysis of SARS-CoV-2 provides better understanding of its epidemiology, pathogenesis and to devise antiviral therapeutic strategies against COVID-19.
The results of our study revealed, a total of 33 mutations identified from 675 sequences of NSP5 (main viral protease) from India. Amongst these mutations, three were non-synonymous amino acid substitutions (N142, L75F and C22N), whereas others showed deleterious impact on the structure and function of NSP5 protein. The mutations K236R, N142L, K90R, A7V, C22N and H246Y showed significant alterations in the secondary structure of NSP5 protein. The mutations N142L, H246Y and I43V were destabilizing and possess -∆∆G values. All NSP5 mutants except for I43V and K90R mutant (positive values) showed negative values of ΔΔSVib ENCoM and hence resulted in rigidification of protein structure. Due to these mutations, considerable alterations were observed at several positions that also affect its stability and dynamicity which in turn altered the function of NSP5. Roe et al29 have reported that NSP5 are capable to make associatation with several other components of replication complex. Earlier studies have also revealed that important intra- and intermolecular interaction exist between the main viral protease NSP5 and other replicase gene, with mutation in the NSP5 domain as well as in the NSP3 and NSP10 which negatively affecting the activity of NSP5.29–31The design and development of vaccine gained much attention nowadays including the multiepitope, DNA as well as RNA-based vaccines for various infectious diseases (such as influenza virus, Ervebo virus), using predictive tools of immunoinformatics have become the major research priority. The conventional methods of vaccine designing strategies include experimental identification, establishing immunological correlation with the coronavirus to develop potential vaccine construct. For the structural activities of SARS-CoV-2, proteins are supposed to be important constituent involved in the viral infection, entry and replication. The findings of earlier studies suggested that protein could be a very good target for developing vaccine against SARS-CoV-2.32–34 Additionally, for a peptide vaccine to be highly immunogenic B-cell epitope of its target molecule must interact with a T-cell immune epitope. The T-cell epitopes is made up of short fragments of peptide and hence appeared as more propitious, which generate long-term immune response mediated by CD8+ T-cells.6 In contrast, the B-cell epitopes consists of lineal chain of amino acid.35,36
The epitope selection based on immunogenic features like antigenicity, allergenicity and toxicity. Similarly, the predicted antigenic determinants (epitopes) of MHC class-I showed interaction with the several HLA alleles and, therefore, found to be antigenic. The hydropathy index and physiochemical properties of SARS-CoV-2 NSP5 protein were also estimated which revealed that protein is stable and can form non-covalent bonds (such as hydrogen bonds) with other protein molecules. The present in silico study was found consistent with the previous studies based on immunoinformatics approach for the design and developments of novel therapeutic intervention and/or vaccine against COVID-19.37–40
In this study, we investigated the NSP5, as a potent immunogenic epitopes which elevates prolonged humoral (B-cell) as well as cell-mediated (T-cell) immune response to counteract viral particles, and hence serves as a potential candidate vaccine. A total of eight B-cell and T-cell epitopes were predicted for NSP5 proteins, amongst which the epitopes KMAFPSGKV, EDMLNPNYEDL, QNGMNG and EFTPFDVVR were highly immunogenic as well as non-allergenic. Primarily, the efficacy of vaccine candidates relies on the selection of its antigen molecules.41 The data obtained from our study also corroborates the previous findings. Earlier studies on SARS-CoV and MERS-CoV have shown that the S glycoprotein can induce antibodies to neutralize virus infection by blocking virus binding as well as its fusion to the host cell.41,42 Yashvardhini et al17 have also reported that multiepitopes-based peptide vaccines are safe and specific that need adjuvants to show high levels of immunogenicity.
In the present study, occurrence of recurrent mutations in the main viral protease (NSP5) of coronavirus elucidates structural alteration that might affect its functions. Using predictive tools of computational biology, we also predicted promising epitope based vaccine candidates that are capable to stimulate both humoral (B-cell) as well as cellular (T-cell) immune responses. However, our in silico designed vaccine construct showed high efficacy and, therefore, suggested as good candidate against SARS-CoV-2 infections. Moreover, further in vivo and in vitro studies are mandatory to validate the durability and efficacy of designed vaccine candidate.
ConclusionOccurrence of recurrent mutations in the NSP5 of SARS-CoV-2 provides a deep insight in the identification and magnitude of virulence properties. The study also suggests continues molecular surveillances of novel coronavirus that might be useful in the development of ongoing biomedical intervention to curb this contagious disease. For the design and development of candidate vaccine, NSP5 of coronavirus has been chosen as potentially ideal target molecule because NSP5 is the main viral protease of SARS-CoV-2 and plays an important role in the viral replication cycle. Moreover, our study sheds light on, high efficacy and durability of designed epitopes vaccine candidate applying various predictive tools of immunoinformatics; further, in vivo and in vitro studies are mandatory to validate designed candidate vaccine.
The following are the supplementary data related to this article.
Supplementary material 1
Supplementary material 2
Supplementary material 3
Supplementary data to this article can be found online at https://doi.org/10.1016/j.vacun.2021.10.002.
Trial registration number (if clinical trial)None.
FundingNil.