







The literature on inducers in epilepsy and bipolar disorder is seriously contaminated by false negative findings. Part II of this comprehensive review on antiepileptic drug (AED) inducers provides clinicians with further educational material about the complexity of interpreting AED drug–drug interactions.
The basic pharmacology of induction is reviewed including the cytochrome P450 (CYP) isoenzymes, the Uridine Diphosphate Glucuronosyltransferases (UGTs), and P-glycoprotein (P-gp). CYP2B6 and CYP3A4 are very sensitive to induction. CYP1A2 is moderately sensitive while CYP2C9 and CYP2C19 are only mildly sensitive. CYP2D6 cannot be induced by medications. Induction of UGT and P-gp are poorly understood. The induction of metabolic enzymes such as CYPs and UGTs, and transporters such as P-gp, implies that the amount of these proteins increases when they are induced; this is almost always explained by increasing synthesis mediated by the so-called nuclear receptors (constitutive androstane, estrogen, glucocorticoid receptors and pregnane X receptors). Although part i provides correction factors for AEDs, extrapolation from an average to an individual patient may be influenced by administration route, absence of metabolic enzyme for genetic reasons, and presence of inhibitors or other inducers. AED pharmacodynamic DDIs may also be important. Six patients with extreme sensitivity to AED inductive effects are described.
La literatura sobre los inductores en los casos de epilepsia y trastorno bipolar está contaminada por falsos negativos. Esta segunda parte de una amplia revisión sobre los fármacos antiepilépticos (FAE) aporta más material educativo a los clínicos acerca de la complejidad de interpretar las interacciones farmacológicas.
Se revisa la farmacología básica de la inducción incluyendo los citocromos P450 (CYP), las enzimas de glucuronización (UGT) y la glucoproteína P (P-gp). CYP2B6 y CYP3A4 son muy sensibles a la inducción. CYP1A2 es moderadamente sensible. CYP2C9 y CYP2C19 son solo levemente sensibles. CYP2D6 no puede ser inducida por los fármacos. La inducción de las enzimas metabólicas, CYP o UGT, y los transportadores como la P-gp, se debe a un incremento de la síntesis de estas proteínas mediado por los denominados receptores nucleares (receptores constitutivo de androstano, de los estrógenos, de los glucocorticoides y de pregnano X). Aunque la primera parte de este trabajo describe los factores de corrección para los antiepilépticos inductores, la extrapolación de estos valores para un paciente promedio a un individuo concreto está influenciada por la ruta de administración, la carencia de la enzima metabólica debida a razones genéticas, y la presencia de inhibidores, u otros inductores. También pueden ser importantes las interacciones farmacológicas de los fármacos antiepilépticos al nivel de los mecanismos farmacodinámicos. Se describen 6 pacientes con una sensibilidad extrema a los inductores antiepilépticos.
The neuropsychopharmacology literature on drug–drug interactions (DDIs) with drug metabolic inducers is seriously contaminated by false negative findings. Inducers’ effects are systematically denied or at least undervalued, and the published literature on antiepileptic drugs (AEDs)1 and on bipolar disorder2 systematically de-emphasizes their clinical relevance. This two-part article provides information for clinicians to compensate for this oversight in the literature on AED inducers. Part I introduced the subject of potent (Table 1) and mild inducers (Table 2) and provided clinical recommendations on correcting the effect of inducers through dose modifications of the induced substrates by using correction factors (Table 3).a As the available literature for calculating these correction factors is seriously limited, the author acknowledges that it is likely that in 5 years this review article may be obsolete and that the correction factors provided may need to be extensively modified.
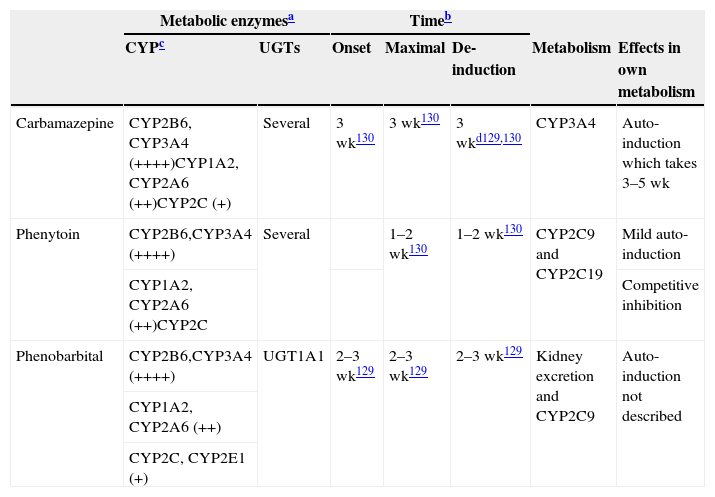
Characteristics of potent inducers.
| Metabolic enzymesa | Timeb | ||||||
|---|---|---|---|---|---|---|---|
| CYPc | UGTs | Onset | Maximal | De-induction | Metabolism | Effects in own metabolism | |
| Carbamazepine | CYP2B6, CYP3A4 (++++)CYP1A2, CYP2A6 (++)CYP2C (+) | Several | 3 wk130 | 3 wk130 | 3 wkd129,130 | CYP3A4 | Auto-induction which takes 3–5 wk |
| Phenytoin | CYP2B6,CYP3A4 (++++) | Several | 1–2 wk130 | 1–2 wk130 | CYP2C9 and CYP2C19 | Mild auto-induction | |
| CYP1A2, CYP2A6 (++)CYP2C | Competitive inhibition | ||||||
| Phenobarbital | CYP2B6,CYP3A4 (++++) | UGT1A1 | 2–3 wk129 | 2–3 wk129 | 2–3 wk129 | Kidney excretion and CYP2C9 | Auto-induction not described |
| CYP1A2, CYP2A6 (++) | |||||||
| CYP2C, CYP2E1 (+) | |||||||
++++: massive induction; ++: moderate induction; +: mild induction. wk: weeks.
These are approximated times provided by review articles.129,130 Readers may need to be aware that few studies have been conducted to verify these times.
Not all CYPs have the same ability to be induced by potent inducers. More details are provided in Table 5. It is believed that potent inducers have massive effects (++++) on CYP2B6 and CYP3A4. On the other hand, potent inducers have only mild to moderate effects on the CYP2C subfamily which includes CYP2C8, CYP2C9 and CYP2C19.131 Although the literature is not specific on this point, the author believes that CYP1A2 may be induced intermediately between the potent effects on CYP2B6 and CYP3A4 and mild effects on the CYP2C subfamily and described as moderate (++). There is limited information on CYP2A6 suggesting potential for moderate induction (++), but clinicians need to be aware that few drugs are metabolized by CYP2A6; this is the main metabolic pathway for nicotine. There is limited information on CYP2E1 which may have mild potential for induction, but clinicians need to be aware that few drugs are metabolized by CYP2E1, although it is a minor metabolic pathway for alcohol and some antiepileptic drugs.
The loss of induction may take longer for CYP1A2 substrates than for CYP3A substrates (respective induction half-lives were 105 and 70h, or 4.4 days and 2.9 days.132
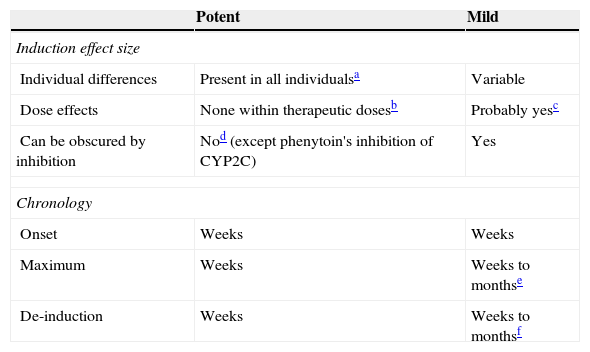
Mild inducers: comparison to potent inducers.
| Potent | Mild | |
|---|---|---|
| Induction effect size | ||
| Individual differences | Present in all individualsa | Variable |
| Dose effects | None within therapeutic dosesb | Probably yesc |
| Can be obscured by inhibition | Nod (except phenytoin's inhibition of CYP2C) | Yes |
| Chronology | ||
| Onset | Weeks | Weeks |
| Maximum | Weeks | Weeks to monthse |
| De-induction | Weeks | Weeks to monthsf |
Although it has not been systematically studied, it is generally accepted that potent inducers tend to maximally induce all patients as long as they are given doses beyond those causing maximal induction.
It is also usually believed that a therapeutic dose for epilepsy should cause maximal induction in most patients. Therefore, further increased doses beyond therapeutic doses may not cause more induction. Similarly, giving another potent inducer to a person taking usual doses of one potent inducer may not make a difference.
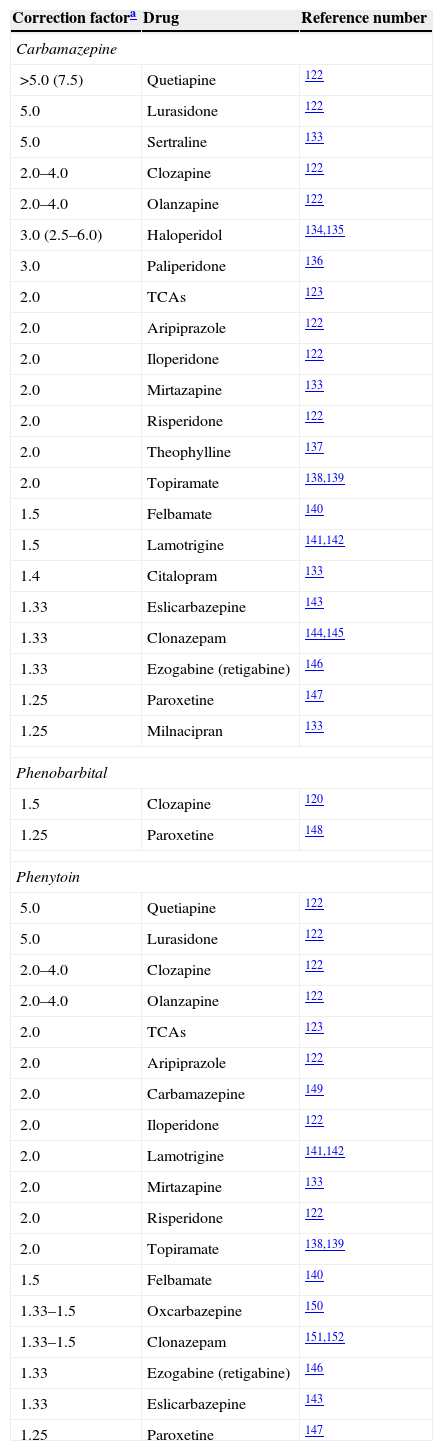
Correction factors for potent inducers.
| Correction factora | Drug | Reference number |
|---|---|---|
| Carbamazepine | ||
| >5.0 (7.5) | Quetiapine | 122 |
| 5.0 | Lurasidone | 122 |
| 5.0 | Sertraline | 133 |
| 2.0–4.0 | Clozapine | 122 |
| 2.0–4.0 | Olanzapine | 122 |
| 3.0 (2.5–6.0) | Haloperidol | 134,135 |
| 3.0 | Paliperidone | 136 |
| 2.0 | TCAs | 123 |
| 2.0 | Aripiprazole | 122 |
| 2.0 | Iloperidone | 122 |
| 2.0 | Mirtazapine | 133 |
| 2.0 | Risperidone | 122 |
| 2.0 | Theophylline | 137 |
| 2.0 | Topiramate | 138,139 |
| 1.5 | Felbamate | 140 |
| 1.5 | Lamotrigine | 141,142 |
| 1.4 | Citalopram | 133 |
| 1.33 | Eslicarbazepine | 143 |
| 1.33 | Clonazepam | 144,145 |
| 1.33 | Ezogabine (retigabine) | 146 |
| 1.25 | Paroxetine | 147 |
| 1.25 | Milnacipran | 133 |
| Phenobarbital | ||
| 1.5 | Clozapine | 120 |
| 1.25 | Paroxetine | 148 |
| Phenytoin | ||
| 5.0 | Quetiapine | 122 |
| 5.0 | Lurasidone | 122 |
| 2.0–4.0 | Clozapine | 122 |
| 2.0–4.0 | Olanzapine | 122 |
| 2.0 | TCAs | 123 |
| 2.0 | Aripiprazole | 122 |
| 2.0 | Carbamazepine | 149 |
| 2.0 | Iloperidone | 122 |
| 2.0 | Lamotrigine | 141,142 |
| 2.0 | Mirtazapine | 133 |
| 2.0 | Risperidone | 122 |
| 2.0 | Topiramate | 138,139 |
| 1.5 | Felbamate | 140 |
| 1.33–1.5 | Oxcarbazepine | 150 |
| 1.33–1.5 | Clonazepam | 151,152 |
| 1.33 | Ezogabine (retigabine) | 146 |
| 1.33 | Eslicarbazepine | 143 |
| 1.25 | Paroxetine | 147 |
Bupropion's correction factor for carbamazepine was 10.0, calculated by the author from the limited information available.133
Unfortunately, it is a major simplification to think that correction factors can completely resolve clinicians’ problems when trying to deal with the complex issue of induction in neuropsychopharmacology. Many of the drug combinations that neurologists and/or psychiatrists find in daily clinical practice are not addressed in Table 3. Part I provides a conservative attempt to reflect the complexity of the issue by describing mild inducers that can also behave as inhibitors (Table 4). Part II is an effort to further educate clinicians about the complex nature of interpreting AED DDIs through the provision of basic pharmacological knowledge to help to improve their ability to interpret complex DDIs.
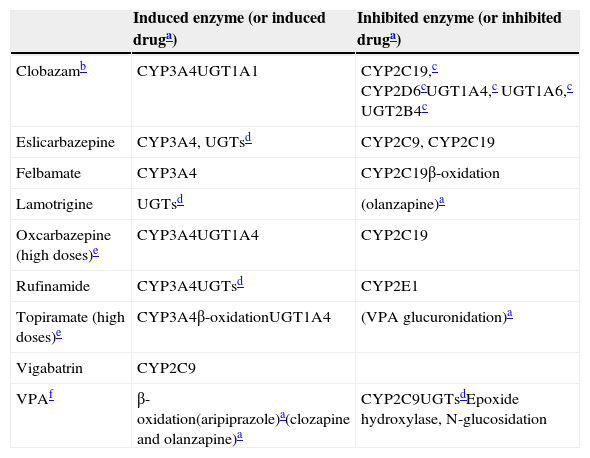
Mild inducers: their inductive and inhibitory properties.
| Induced enzyme (or induced druga) | Inhibited enzyme (or inhibited druga) | |
|---|---|---|
| Clobazamb | CYP3A4UGT1A1 | CYP2C19,c CYP2D6cUGT1A4,c UGT1A6,c UGT2B4c |
| Eslicarbazepine | CYP3A4, UGTsd | CYP2C9, CYP2C19 |
| Felbamate | CYP3A4 | CYP2C19β-oxidation |
| Lamotrigine | UGTsd | (olanzapine)a |
| Oxcarbazepine (high doses)e | CYP3A4UGT1A4 | CYP2C19 |
| Rufinamide | CYP3A4UGTsd | CYP2E1 |
| Topiramate (high doses)e | CYP3A4β-oxidationUGT1A4 | (VPA glucuronidation)a |
| Vigabatrin | CYP2C9 | |
| VPAf | β-oxidation(aripiprazole)a(clozapine and olanzapine)a | CYP2C9UGTsdEpoxide hydroxylase, N-glucosidation |
VPA: valproic acid.
For drugs in parentheses, the enzyme behind the induction or inhibition is not definitively established.
Although this second part has much more theoretical information than the first part, the author has selected the information according to his experience with the deficiencies of the literature for teaching clinicians how to navigate the turbulent waters of DDI with inducers in neuropsychopharmacology. Using this practical approach, the author presents seven sections that address three issues: basic pharmacology, the true pharmacological complexity of DDI, and the existence of individuals who are unusually sensitive to induction.
The basic pharmacology of induction reflects the increased production of the proteins involved in pharmacokinetic mechanisms. Inducers increase the metabolism of many neuropsychopharmacological drugs metabolized by the Cytochrome P450 (CYP) isoenzymes, the major oxidative enzymes. Inducers sometimes increase the activity of other less well understood metabolic enzymes, the Uridine Diphosphate Glucuronosyltransferases (UGTs), which are the most important conjugative enzymes. Only very recently has it become clear that, besides metabolizing enzymes, another major group of pharmacokinetic proteins called transporters, which usually work in tandem with metabolic enzymes to eliminate xenobiotics from the body, can also be induced. P-glycoprotein (P-gp) is the most important transporter and is briefly described. A new rapidly expanding area in the literature, describing nuclear receptors, is beginning to provide some understanding of how inducers increase the activity of CYPs, UGTs and P-gp.
In summary, basic pharmacology accounts for the first four sections: (1) CYPs, (2) UGTs, (3) P-gp, and (4) nuclear receptors. The fifth and six sections address the second issue: the complexity of interpreting induction in patients on a polypharmacy regimen (which is the norm in patients with epilepsy and bipolar disorder). They describe, respectively, that inducer effects need to be understood in the context of the complexity of pharmacokinetic polypharmacy and pharmacodynamic DDIs. The seventh section does not come from the literature, but from the author's clinical practice. It acknowledges that, on rare occasions, clinicians find patients who are very sensitive to induction and may require massive dose increases of some drugs.
CYPsCYP terminology can be confusing for clinicians who need to know that CYP names include a number for the family, a letter for the subfamily and then another number for the specific isoenzyme. The first three CYP families are mainly located in the liver and are involved in the metabolism of xenobiotics such as medications.3 They are also important in the activation and deactivation of carcinogens, and they may have some not well-understood role in endogenous metabolism.3 The CYP families higher than “3” appear to be mainly involved in the endogenous metabolism of complex molecules including cholesterol and its derivate, the steroids.3
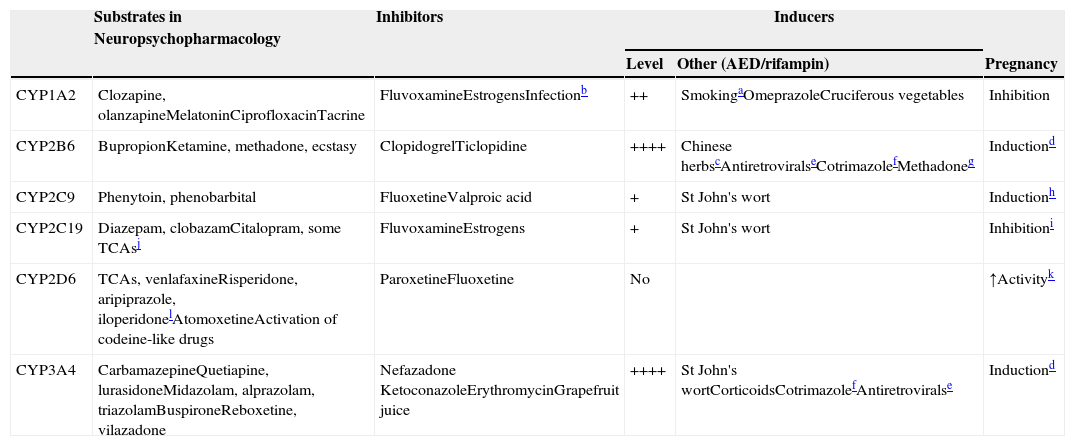
The first 3 CYP families are part of the oxidative enzymes (traditionally called Phase I metabolic enzymes). CYP34A4 is the most important hepatic CYP, accounting for more than one-third of hepatic CYPs. Five other hepatic CYPs, CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP2D6, are definitively important for the metabolism of neuropsychopharmacological drugs (Table 5), and those prescribers not familiar with them need to make a major effort to learn their names and separate one from the other, in spite of their confusing names.
CYPs involved in drug metabolism.
| Substrates in Neuropsychopharmacology | Inhibitors | Inducers | |||
|---|---|---|---|---|---|
| Level | Other (AED/rifampin) | Pregnancy | |||
| CYP1A2 | Clozapine, olanzapineMelatoninCiprofloxacinTacrine | FluvoxamineEstrogensInfectionb | ++ | SmokingaOmeprazoleCruciferous vegetables | Inhibition |
| CYP2B6 | BupropionKetamine, methadone, ecstasy | ClopidogrelTiclopidine | ++++ | Chinese herbscAntiretroviralseCotrimazolefMethadoneg | Inductiond |
| CYP2C9 | Phenytoin, phenobarbital | FluoxetineValproic acid | + | St John's wort | Inductionh |
| CYP2C19 | Diazepam, clobazamCitalopram, some TCAsj | FluvoxamineEstrogens | + | St John's wort | Inhibitioni |
| CYP2D6 | TCAs, venlafaxineRisperidone, aripiprazole, iloperidonelAtomoxetineActivation of codeine-like drugs | ParoxetineFluoxetine | No | ↑Activityk | |
| CYP3A4 | CarbamazepineQuetiapine, lurasidoneMidazolam, alprazolam, triazolamBuspironeReboxetine, vilazadone | Nefazadone KetoconazoleErythromycinGrapefruit juice | ++++ | St John's wortCorticoidsCotrimazolefAntiretroviralse | Inductiond |
++++: massive induction; ++: moderate induction; +: mild induction. AED: antiepileptic drug.
Polycyclic aromatic hydrocarbons in smoke have inductive effects. These compounds also found on char grilled food and coffee from roasted coffee beans, which can also have inductive effects.22
Respiratory infections, other serious infections, such as pyelonephritis or appendicitis or even major inflammations can inhibit CYP1A2 because the cytokines released inhibit CYP1A2.
Sodium ferulate was an inducer of bupropion metabolism in a study. It is the sodium salt of ferulic acid, which is widely distributed in herbs and Chinese formulas such as Ligusticum, Chuanxiong and Chaihu–Sugan–San.14 Another inducer is baicalin, a flavone glucuronide extracted from the medical plant Radix scutellariae, which is present in fruits, vegetables, and beverages derived from plants (tea, red wine), and in a wide range of herbal medicines including Huang-Lian-Jie-Du-Tang, hangeshashinto, San-Huang-Xie-Xin-Tang, Da-Chai-Hu-Tang, and Xiao-Chai-Hu-Tang.15
Pregnancy definitively induces CYP2B6 and CYP3A4.25 According to an in vitro study, CYP2B6 and CYP3A4 are induced by both estrogen and progesterone. Progesterone also induces CYP3A5.
In vitro studies indicate that methadone may induce its own metabolism and this may be mediated not only by CYP2B6 but also by CYP3A4.20
It is believed that CYP2C9 increases during pregnancy because phenytoin clearance increases. It cannot be ruled out that mechanisms other than CYP2C9 induction may explain changes in phenytoin clearance during pregnancy. Estradiol increases the activity of CYP2C9 without affecting expression by unknown mechanisms.26
Estrogens are thought to be competitive inhibitors of CYP2C19, but a recent study suggested that they may inhibit CYP2C19 expression.12
CYP2C19 is the main enzyme to demethylate amitriptyline, clomipramine and imipramine. Their metabolites then are further metabolized by hydroxylation, mainly by CYP2D6.
It is not well understood why CYP2D6 activity may increase in pregnancy, since it is believed that CYP2D6 cannot be induced. A recent study suggested that pregnancy may remove a suppressor of CYP2D6 expression.24
There are multiple extra-hepatic CYP isoenzymes involved in xenobiotic metabolism not included in Table 5, but they are less well understood and less important. The two most important extra-hepatic CYPs may be CYP3A4 and CYP3A5. CYP3A4 is also the most important CYP in the small intestine.4 The small intestine CYP3A4 (with P-gp; see that section) is a fundamental component of what pharmacologists usually call first-pass metabolism. First-pass metabolism5 refers to the substantial decrease in absorption of some drugs when using the oral versus the intravenous (IV) route of administration. Metabolism in the intestine and the first time it goes though the liver constitutes first-pass metabolism. Although some drugs displaying first-pass metabolism are metabolized by UGTs (e.g., asenapine), there is agreement that CYP3A4 (and P-gp) located in the intestine and liver explain first-pass metabolism for CYP3A4 substrates. CYP3A5 has high homology with CYP3A4, appears to metabolize many of the same substrates as CYP3A4, and is particularly important in kidney drug metabolism. In general, for the majority of CYP3A drugs, the contribution of CYP3A5 to their total metabolism is thought to be small.6
The substrates column in Table 5 describes which drugs in neuropsychopharmacology7,8 are metabolized by the five most important hepatic CYPs. Another column describes the CYP inhibitors.9–13 Inhibitors usually act by binding to the CYP, making that CYP molecule inactive. The table focuses on potent inhibitors that cannot easily be displaced by substrates, but any substrate in the right clinical circumstances can behave as a clinically relevant competitive inhibitor of its drug-metabolizing enzymes. Some CYP inhibitors, particularly the female sexual hormones, may not only inactivate some CYPs (CYP1A2 and CYP2C19) but may also decrease their synthesis.12
Table 5’s third column describes the level of induction, which refers to how sensitive the CYP is to induction (see the section on complex pharmacokinetic polypharmacy). The next column lists other inducers9,14–22 besides AEDs (see Tables 1 and 4). Rifampin is not listed for all five CYPs since it is a promiscuous inducer of these five CYPs (and other enzymes). The last column focuses on the complex effects of pregnancy, which can increase or decrease the activity of these CYPs.12,23–26
Genetic variationsThe first three CYP families include genes that are highly polymorphic, meaning that genetic variations affecting their function are frequent. Although it is not well understood, the genetic variations of the first CYP families may be related to different evolutionary pressures from environmental differences in exposure to different xenobiotics, particularly in the diet. It is likely that our ancestors, in times of hunger, had no choice but to eat whatever plants they could find to palliate their hunger. Many plants have toxins on them to protect them from plant-eating animals. As most medications are derived from plants, it is not surprising that we use the same CYPs to metabolize medications. Clinicians need to know that the first three CYP families vary from species to species; thus, CYP animal studies do not extrapolate well to humans and in vitro studies with human hepatocytes are used to study these first three CYPs in the laboratory.
Clinicians are frequently bombarded by companies marketing the benefits of CYP genotyping. The author thinks that to interpret these genotyping results clinicians need a sophisticated knowledge of CYP alleles and the technical limitations of the various genotyping techniques.27–29
As summarized in Table 6, the author's opinion is that most CYP variations are not ready for clinical use.29 Only three of them, CYP2C9, CYP2C19 and CYP2D6, may be ready, but CYP2C9 has relatively small applications in neuropsychopharmacology. CYP1A2,30,31 CYP2B6,32,33 CYP3A4,34 and CYP3A534,35 genotyping are described as limited since we have very limited understanding of their genetic variations and the genotypic-phenotypic relationships. It is easy to find articles marketing the potential of CYP genotyping by stressing the studies with significant results but ignoring the studies with non-significant results and major gaps in understanding the genotype-phenotype relationships. In the other extreme, one can find investigators with an evidence-based approach who “undermine” CYP genotyping by explaining that it has very little role in selective serotonin reuptake inhibitor (SSRI) prescription. The small role of CYP genotyping for many SSRIs dosing has little relevance for the field, since any pharmacologist working on SSRIs would have predicted it using pharmacological mechanistic knowledge without the need for any systematic review of the literature.36 Regardless, recommendations for dose corrections according to CYP2C19 genotyping have been provided for citalopram, escitalopram and sertraline.29
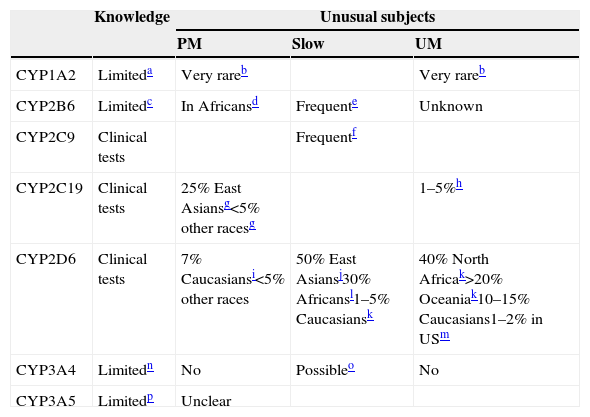
CYP involved in drug metabolism: genetic variations.
| Knowledge | Unusual subjects | |||
|---|---|---|---|---|
| PM | Slow | UM | ||
| CYP1A2 | Limiteda | Very rareb | Very rareb | |
| CYP2B6 | Limitedc | In Africansd | Frequente | Unknown |
| CYP2C9 | Clinical tests | Frequentf | ||
| CYP2C19 | Clinical tests | 25% East Asiansg<5% other racesg | 1–5%h | |
| CYP2D6 | Clinical tests | 7% Caucasiansi<5% other races | 50% East Asiansj30% Africansl1–5% Caucasiansk | 40% North Africak>20% Oceaniak10–15% Caucasians1–2% in USm |
| CYP3A4 | Limitedn | No | Possibleo | No |
| CYP3A5 | Limitedp | Unclear | ||
≈: approximately; PM: poor metabolizer. UM: ultrarapid metabolizer. AA: African Americans.
a Some studies indicate that some alleles may influence induction. CYP1A2*1C has been associated with low inducibility and CYP1A2*1F with high inducibility but there is no complete agreement in the literature.30,31
b CYP1A2 review articles31 usually report that there are no clearly identified PMs with missing enzyme or UMs with clearly increased activity. Rarely, clozapine literature describes some extreme subjects compatible with being CYP1A2 PM (e.g., a patient with one CYP1A2*7 allele41). Some cases with a UM profile have been described but no clear genetic variants have been identified.42,43
c CYP2B6 activity is associated with a large number of CYP2B6 genetic variations including single nucleotide polymorphisms and haplotypes.33
d CYP2B6*18 occurs predominantly in Africans and does not express a functional protein, at least for some substrates. Frequencies of individuals with only one deficient allele are described as 5–12% of Africans and 4–8% of African-Americans.14 Thus, although PM frequencies are not described in articles, PM frequencies should be expected to be <1% in the general population.
e CYP2B6*6 is probably the most frequent low functioning allele and occur 15% to over 60% in different populations.32
f Several frequent CYP2C9 alleles have lower activity; CYP2C9*1 is the normal allele and a subject with *1/*1 will have an activity of 1.0. CYP2C9*2 has lower activity, according to Castellan et al.20; a subject with *2/*2 will have an activity of 0.70 while a subject with *1/*2 will have an activity of 0.82. CYP2C9*3 has very low activity, according to Castellan et al.40 a subject with *3/*3 will have an activity of 0.13 while a subject with *1/*3 will have an activity of 0.56 and *2/*3 will have an activity of 0.39. Therefore, for *3/*3 a dose reduction of 65% of phenytoin and 85% of phenobarbital is recommended.
g For CYP2C19, the most important inactive alleles are *2 and *3, which are frequent in East Asians.38
h CYP2C19*17 is associated with increased activity. Scott et al. (2011)52 described 21% of Europeans and 18% of Africans as having one CYP2C19*17 allele. With these frequencies, it can be estimated that approximately 4% of Europeans and 3% of Africans would have two alleles with increased activity and may be considered CYP2C19 UMs (CYP2C19*17/CYP2C19*17). Other authors argue that CYP2C19*17 may be irrelevant for the majority of drugs.53
i The most important CYP2D6 inactive alleles are *3, *4, *5 (deletion) and *6.38
Different review articles provide different prevalences of CYP2D6 PMs. In a recent review, McGraw and Waller45 described 3–10% in Caucasians, 0–19% in African Americans, 6.0% in Hispanics, 0–4.4% in Native Americans and 0.3% in East Asians.
j In CYP2D6, the slow metabolizers are usually called intermediate metabolizers (IMs), but the terminology varies from laboratory to laboratory. Sometimes CYP2D6 tests describe subjects with at least one normal allele as intermediate metabolizers while others will consider that an EM.27,38 East Asians frequently have *10. Assuming that two normal active alleles have an activity of 1, one active allele and *10 have an activity level of 0.54 and two *10 only 0.10.49
k According to Tod et al.,49 typical CYP2D6 UMs with 3 active alleles (1 normal allele in one chromosome and duplication in the other) have an activity level of 1.67 (1.15–2.21). A comprehensive worldwide study provided these frequencies.54
l CYP2D6*17 is found in individuals with African ancestry and usually has lower activity, but has normal activity for risperidone.37
m In a large study in a U.S. psychiatric hospital,37 the prevalence of CYP2D6 UMs was 1.5% (95% confidence interval, CI, 1.1, 1.9). In African Americans it was 2.0% (95% CI 1.1, 3.7) for many drugs but as CYP2D6*17 may have normal activity for risperidone, the frequency of UMs for risperidone was slightly higher at 2.9% (CI 1.7, 4.9).
n CYP3A4 expression and activity greatly vary interindividually, but it is believed that it is due not only to genetic factors but to nongenetic factors such as hormone and health status, and the impact of environmental stimuli. Most of the described genetic variants have no functional consequences or are too rare to contribute significantly to the CYP3A4 variability.34
o A new allele CYP3A4*22, associated with low activity, has been described in 5–7% of Caucasians.50 It may have clinical relevance for CYP3A4 drugs such as quetiapine.51
p CYP3A5 literature is rather confusing. The traditional story is that there are 3 alleles with no or low expression: CYP3A5*3, CYP3A5*6 and CYP3A5*7. CYP3A5*6 and CYP3A5*7 are only found in people with African ancestry. Individuals are usually classified as high and low CYP3A5 expressors. Approximate frequencies of high expressors are 70% in Africans and 10% in Caucasians.34 More recently, Bains et al.35 reported that it is not definitively established that CYP3A5*6 is associated with lower expression; even assuming that the number of high expressors in African populations varies widely, it may average 43%. To make the CYP3A5 story more confusing, the relevance of CYP3A5 polymorphism may vary from drug to drug and is not definitively established.34
Therefore, despite the author's genotyping of more than 5000 patients with CYP2D6 and/or CYP2C19,37 he thinks that CYP genotyping should be used judiciously in specific individuals with unusual responses that would be compatible with CYP2D6 and/or CYP2C19 genetic abnormalities. This genotyping should be combined with information on inhibitors and inducers. In the author's experience, CYP2D6 and CYP2C19 genotyping can occasionally be useful in patients with peculiar response to some psychiatric drugs,38 particularly first-generation psychiatric drugs, since tricyclic antidepressants (TCAs) and phenothiazine antipsychotics were mainly metabolized by CYP2D6 and have narrow therapeutic windows. As a matter of fact, excellent TCA dosing guidelines based on CYP2D6 and CYP2C19 genotyping have been developed.7 With second-generation antipsychotics and antidepressants, CYP genotyping is less helpful, since they have very different metabolic pathways.9,13,39 Recommendations for dose corrections according to CYP2D6 genotyping have been provided for venlafaxine, aripiprazole, risperidone, zuclopenthixol and atomoxetine.29 The author is frequently consulted by patients and colleagues regarding some extreme subjects who do not tolerate or are not responding to many drugs from the same class, but these situations are not usually explained by CYP genetic variations but by other factors.38
The most important genetic variations in the first three CYP families are described in Table 2.27,31,32,37,38,40–54 The terminology of CYP genotyping and phenotyping is pretty confusing for clinicians. Its complexity is due to the complex historical development of CYP knowledge.55 First, the concept of poor metabolizers (PMs) was described. It was clear that for CYP2D6 and CYP2C19 there were some subjects that appeared to have very low metabolic activity. One of the first descriptions referred to TCAs; some subjects had very low ability to metabolize TCAs.55 Later, it was discovered that TCAs were metabolized by CYP2D6; CYP2D6 PMs are described as those who do not have any CYP2D6 in their livers. Each has two inactive CYP2D6 alleles (one inactive allele in one chromosome from the father and another inactive allele in the other chromosome from the mother), such that no CYP2D6 protein is produced or, if it is produced, it is completely inactive. CYP2D6 phenotypes included PMs, who do not have CYP2D6 in their bodies, and normal individuals who were called extensive metabolizers (EMs). The CYP2D6 population's phenotype was described as bimodal, one mode for PMs and another for EMs. Later, some individuals from one family in Sweden were discovered to have undetectable TCA levels despite taking TCAs; they had more CYP2D6 than normal.56 They had from 3 to 13 active CYP2D6 alleles. The patient with 3 alleles received 1 normal allele from one parent and 2 active alleles from the other parent (the allele had been duplicated). The patient with 13 alleles received 1 normal allele from one parent and 12 active copies from the other parent (the allele had been multiplied 12 times).
CYP2C19 was also discovered to be polymorphic with PMs and EMs55 but has a different racial distribution than CYP2D6 (Table 6). Later on, CYP2C19 UMs were also described; they have an allele that is associated with higher CYP2C19 expression. Thus, genetic mechanisms explaining UMs can include multiplication of an active gene (this is usually included in the concept of copy number variation) such as in CYP2D6, or an allele with increased expression, such as in CYP2C19.
Clinicians need to remember that an individual with an EM genotype can have a PM phenotype when he/she is taking a powerful inhibitor that eliminates all the activities of that enzyme. One can have a UM phenotype when he/she is taking a powerful inducer that increases the activity of any relevant enzyme.
In CYP2C9, no PMs were found but subjects with low activity were found; they were sometimes called slow metabolizers. Some subjects, particularly East Asians, were found to have CYP2D6 present with low activity. They were called intermediate metabolizers (IMs). Therefore, for CYP2D6 and CYP2C19, the genetic variations vary from race to race and include subjects on a continuum from no or low activity to those with excessive activity (Table 6).
Some very rare subjects, not well studied in the literature, are relevant for prescribers in neuropsychopharmacology; they are the “double” PMs for both CYP2D6 and CYP2C19.57 This combined genetic variant is very rare (<1/1000) but may be very relevant for antidepressant prescription. Most oral antidepressants are metabolized by CYP2D6 and/or CYP2C19; these rare double PMs, in the experience of the author, have very negative experience with most antidepressants since they cannot tolerate them well in normal doses. However, they should be able to tolerate normal doses of mirtazapine, which is not dependent on CYP2D6 or CYP2C19 for its metabolism57 or normal doses of bupropion which is mainly metabolized by CYP2B6.
Clinicians need to be aware that a new genetic area relevant for CYP function has recently been receiving attention, but its clinical relevance is not well established. The CYP oxidoreductase (POR) is a microsomal enzyme closely located near the various CYPs; it influences CYP function by donating electrons. It has recently been proposed that POR genetic variations may be better predictors of CYP3A4 phenotype than CYP3A4 genetic variations58 and that POR genetic variants can be associated with increased or decreased activity of CYPs from the first three families.59
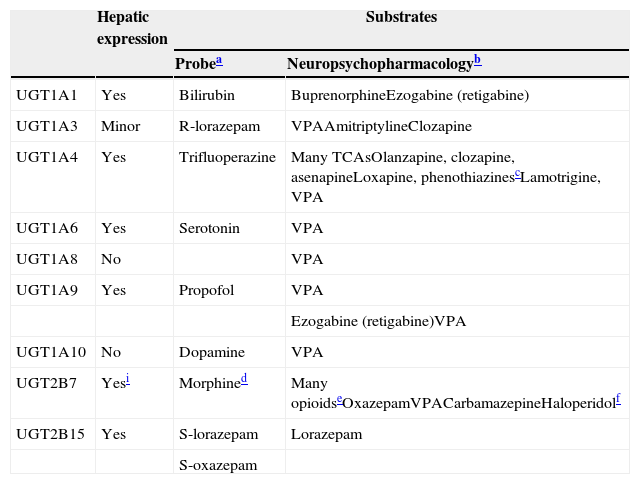
UGTsUGTs are the most important of the conjugation enzymes (traditionally called Phase II metabolic enzymes) and are the main metabolic enzymes for a few antipsychotics, a few AEDs, and a few benzodiazepines (Table 7). UGTs have been neglected and are less well understood than CYPs.60 Knowledge of UGTs is 10–15 years behind CYPs, to the point that the information provided in this review of UGTs on inducer effects is much less reliable than that of CYP inducers and much more likely to need corrections and updates in 5–10 years. Factors contributing to this neglect include: (1) the difficulty of developing analytic methods to measure glucuronides; (2) the overlapping activity of UGTs and the lack of selective probes; (3) the complexity of the glucuronidation cycle, comprising reabsorption throughout the enterohepatic cycle and participation in deconjugation by β-glucuronidases, including those present in bacteria; and (4) endogenous compounds that are frequently substrate, inhibitor or inducer UGTs; this occurs more frequently than in CYPs, providing an additional level of complexity in DDIs.61,62
UGTs involved in drug metabolism.
| Hepatic expression | Substrates | ||
|---|---|---|---|
| Probea | Neuropsychopharmacologyb | ||
| UGT1A1 | Yes | Bilirubin | BuprenorphineEzogabine (retigabine) |
| UGT1A3 | Minor | R-lorazepam | VPAAmitriptylineClozapine |
| UGT1A4 | Yes | Trifluoperazine | Many TCAsOlanzapine, clozapine, asenapineLoxapine, phenothiazinescLamotrigine, VPA |
| UGT1A6 | Yes | Serotonin | VPA |
| UGT1A8 | No | VPA | |
| UGT1A9 | Yes | Propofol | VPA |
| Ezogabine (retigabine)VPA | |||
| UGT1A10 | No | Dopamine | VPA |
| UGT2B7 | Yesi | Morphined | Many opioidseOxazepamVPACarbamazepineHaloperidolf |
| UGT2B15 | Yes | S-lorazepam | Lorazepam |
| S-oxazepam | |||
a Probes are drugs that are used to study the specific enzyme since they will be relatively specific substrates, but for UGTs different articles provide different probes. Only one recent article by an expert in the area73 was used to develop this column.
b Updated from de Leon60 by using de Leon et al.74
c Chlorpromazine and trifluoperazine are phenothiazines that definitively are UGT1A4 substrates.
d Morphine is metabolized to a 3-glucuronide with no analgesic effect by UGT2B7, UGT1A8, and UGT1A3 and to a 6-glucuronide that is more potently analgesic than morphine, mainly by UGT2B7.75
e Many opioids are glucuronized by UGT2B7 besides morphine, including buprenorphine, codeine, naloxone and naltrexone. Buprenorphine and naltrexone can also by glucuronized by UGT1A1.60
f A recent in vitro study76 indicated that haloperidol is mainly glucuronized by UGT2B7 with minor contributions of UGT1A4 and UGT1A9. These UGTs would be responsible for 50–60% of haloperidol clearance versus 20–30% from CYP3A4.
UGTs are located in the internal membrane facing the luminal side of the endoplasmic reticulum of hepatic and extra-hepatic tissue (particularly skin, lung, small intestine and kidney) with direct access to the metabolites resulting from CYPs and oxidizing enzymes.63 More recently, some authors have proposed that protein-protein interactions may be important in understanding UGT function. UGTs may work by forming complexes of several UGTs with glucuronidate substrates at the same time.62 If that idea is proven correct, it would make no sense to try to establish which specific UGT may be important in metabolizing a specific substrate. Moreover, there is some limited information that CYPs and UGTs may interact to metabolize drugs62 and that UGT function may be crucial in understanding the physiological properties of many CYP inhibitors.64
UGTs transfer the glucuronyl group from uridine-5′ diphosphoglucuronate to many lipophilic compounds. The resulting glucuronide is more water soluble, less toxic, and more easily excreted than the parent compound. Glucuronides account for most of the detoxified material found in bile and urine. UGTs have evolved to catalyze the glucuronidation of both endogenous compounds (bilirubin, thyroid hormones, sexual hormones, and serotonin) and xenobiotics (e.g., acetaminophen and morphine are predominantly cleared in this way). The biological importance of glucuronidation is grossly underinvestigated, although it seems reasonable to postulate that it serves primarily to enhance the elimination of substrates from the body,65 but in some instances, it may produce more active or more toxic compounds.65,66
Human UGT genes are classified into four families based on sequence identity: the UGT1 family glucuronidates bilirubin and xenobiotic phenols; the UGT2 family glucuronidates steroids and bile acids. The function of the UGT3A family is not known, whereas the UGT8A family has only one human gene that may be involved in membrane biosynthesis.67 Like the CYPs, each species appears to have its own UGTs with various substrates.
The human UGT1 subfamily is derived from a single gene with at least 13 individual promoters/first exons and a shared set of 2–5 exons.67 The messenger RNA encoding each UGT isoform is formed by the fusion of one type of exon 1 to the four exons 2 to 5. Gene mutations in the common exon 2 to 5 regions can lead to changes in activity and/or expression of all isoforms, while gene mutations in the unique exon 1 or promoter region may only affect the unique isoform involved.68 Thus, several potential transcripts can be generated from the UGT1A locus containing unique 5′ ends and identical 3′ ends, including 4 enzymes located in the liver (UGT1A1, UGT1A3, UGT1A4 and UGT1A9) and three less-understood enzymes (UGT1A7, UGTA18 and UGT1A10) with no hepatic expression, but mainly expressed in gastrointestinal epitheliums.69Table 760,70–76 provides a description of the UGTs that are relevant for neuropsychopharmacology.
The UGT2 family has subfamilies that are synthesized from a series of similar genes located in a cassette on chromosome 4. Three olfactory-specific isoforms are included in the UGT2A subfamily. The UGT2B subfamily includes metabolic enzymes that are responsible for the glucuronidation of steroids and bile acids. UGT2B has 12 members, five pseudogenes (inactive genes) and seven genes. Table 7 describes the two best known, UGT2B7 and UGT2B15.
Genetic variationsUGT enzymes can be polymorphic but are not understood well enough to develop a clear table such as the one for CYPs. UGT1A1 polymorphisms are the best understood. Gilbert's syndrome is a genetic variant associated with reduced levels of UGT1A1 that manifests as non-hemolytic unconjugated hyperbilirubinemia without hepatic structural damage.77
Many polymorphic variations in other UGTs besides UGT1A1 have been identified69,78 but in the majority of cases it is not well established whether these variations are functional or not and frequently the associated findings with pharmacological changes have not been replicated. Other key genomic processes such as copy number variations, epigenetic factors and splicing mechanisms may be important for UGT genes, too.79 In summary, no other UGT genetic polymorphism besides UGT1A1, which has been recommended for some anti-cancer drugs, is likely to be widely used in the clinical environment in the next 5 years and clinicians do not need to be familiar with them.
InhibitorsVPA can be used as an example of our limited understanding of glucuronidation physiology and inhibition. The acyl glucuronide conjugate (valproate-glucuronide or VPAG) appears to be the major VPA urinary metabolite and accounts for 30–50% of the VPA dose.80 Five liver UGTs, UGT1A3, UGT1A4, UGT1A6, UGT1A9, and UGT2B7 and the primarily intestinal UGTs, UGT1A8 and UGT1A10,80,81 may be involved in VPA glucuronidation. According to an in vitro study by Argikar and Remmel,80 UGT2B7 has the highest metabolic activity for VPA; with UGT1A6 second, and then the rest of the UGTs have similar activity. Argikar and Remmel80 warned that we do not know how to extrapolate the clinical relevance of extra-hepatic UGTs, such as UGT1A8 and UGT1A10, due to the lack of data on their expression. Besides being metabolized by UGTs, VPA appears to be a clinically relevant inhibitor of some of these UGTs. However, it is not easy to definitively establish through a literature review which UGTs are inhibited by VPA.
In the neuropsychopharmacology literature there is definitive agreement that VPA inhibits the glucuronidation of two drugs, lamotrigine74,80,82 and lorazepam.74,83 They are mainly metabolized by UGTs but, because there is no agreement on which UGTs are inhibited by VPA, this inhibition cannot be explained. If one agrees that UGT1A4 is the most important enzyme in lamotrigine metabolism and UT2B15 for lorazepam, then VPA is probably a clinically relevant inhibitor of UGT1A4 and UGT2B15. Other articles describe VPA as an inhibitor of UGT2B7,84,85 UGT2B15 and UGT1A9.84 In summary, the author is not absolutely sure which UGTs may be inhibited by VPA; the literature provides a complicated and sometimes contradictory picture. The literature does not address whether drug inhibition by VPA during an in vitro study is a sign of inhibition of a specific UGT or inhibition of multiple UGTs. Moreover, it is not clear to the author how an in vitro study using VPA as an inhibitor can be extrapolated to an in vivo case in which not only VPA but VPA metabolites are present, probably contributing to UGT inhibition, and possibly induction, as Part I of this review article indicates. In summary, we have very limited understanding of UGT inhibition (and induction).
P-gpThe relevance of transporters for drug metabolism and DDI has only recently begun to be understood, but understanding has rapidly expanded in the last ten years. Although some authors have suggested that transporters should be considered as phase III of drug metabolism, this terminology is rarely followed.86 Functionally, transporters are classified according to those mediating the uptake of drugs into cells (uptake or influx transporters) and those mediating the export of drugs or drug metabolites out of cells (efflux transporters).87
P-gp is the best-studied drug transporter, but a psychiatrist or neurologist reviewing the literature will find it confusing from the start, since the articles use three different names.88 P-gp was initially described as a surface glycoprotein modifying the permeability to colchicine in an animal cell model. Then it was found to be overexpressed in tumor samples and was associated with resistance to multiple drugs; the gene was called multidrug resistant protein 1 (MDR1) gene. Its high homology with bacterial transporters suggested that P-gp is an efflux transporter. Immunohistochemical studies demonstrated P-gp expression in tissues with secretory or excretory functions (liver, kidney, and gastrointestinal tract) and at blood-tissue barrier sites, such as the blood-brain barrier (BBB). This pattern of expression indicated that P-gp may influence xenobiotic response and toxicity. P-gp was found to be part of a very large family of ATP-binding transporter genes, some of which are involved in human genetic disorders,86 and was renamed ATP-binding cassette sub-family B member 1 (ABCB1). Therefore, articles on this subject may use three headings for the same protein or gene: P-gp, MRD1 or ABCB1.
P-gp plays a central role in the absorption, distribution and excretion of a wide variety of drugs. Pg-p is expressed in several tissues including intestine, kidney, liver, brain, and placenta.87 It acts as a natural defense mechanism against several drugs by limiting their absorption from the gut and penetration into the brain, and promoting their elimination in the bile and urine. P-gp is expressed in the apical membrane of the entire intestine from duodenum to rectum, with a high expression in enterocytes of the small intestine, thereby contributing to a reduced bioavailability of multiple drugs that are substrates of this transporter. In hepatocytes P-gp is located in the canalicular membrane and in the kidney at the luminal side of proximal tubule epithelial cells mediating the export of xenobiotics into bile and urine, respectively. At the BBB, P-gp is one of the main transporters and is particularly located at the luminal endothelial cell, but is present in other cells including neurons and astrocytes.89 The P-gp role at the BBB is very poorly understood and includes substrates with a very different structure that binds to more than one site of action.90
The ABCB1 gene is located on chromosome 7, just as CYP3A4; they appear to have similar regulations and site actions and thus they appear to share the same substrates and inhibitors, although they may have different affinity for them. It is possible that P-gp may be induced, too, and P-gp overexpression may result in the affected drug having limited gastrointestinal absorption or reduced penetration to the brain, or enhanced elimination in the bile and urine. At the gut, both CYP3A4 and P-gp appear to work in tandem, decreasing the absorption of their common substrate and contributing to what was called first-pass metabolism (see CYP section).
Genetic variationsUGT literature is slowly moving to the same type of genetic terminology as the CYP literature. However, the articles on ABCB1 genetic variations are much more confusing. They usually focus on single nucleotide polymorphisms (SNPs) or haplotypes. A SNP is a DNA sequence variation occurring when a single nucleotide (A adenine, T, thymine, C cytosine or G, guanine) varies. A haplotype is a combination of alleles at adjacent locations on a chromosome that are inherited together. The lack of use of numeric alleles denotes that it is not clear which are associated with functional variations in P-gp expression and which are not.91 This is why it is not surprising that attempts to complete meta-analysis of the result of associations between ABCB1 genetic variations and AEDs92 or second-generation antipsychotics are both negative.93 The existence of many significant findings which are rarely replicated suggests to the author that ABCB1 genetic variations will not reach clinical practice in the next five years.
Drug interactions in generalSome articles87 classify P-gp substrates into two groups: (1) the majority of drugs, which are substrates of both P-gp and CYP3A4, and (2) those specific to P-gp, which have no CYP metabolism. The latter include a few drugs such as dabigatran (an oral anticoagulant), digoxin and fexofenadine (an antihistaminic). It is also believed that not all CYP3A4 substrates are P-gp substrates; midazolam is a CYP3A4 substrate but not a P-gp substrate.94
Quinidine and clarithromycin have been demonstrated to be inhibitors of digoxin and this inhibition is thought to be mediated on the basis of potent P-gp inhibition.94,95 However, quinidine is also a potent CYP2D6 inhibitor and clarithromycin a potent CYP3A4 inhibitor. Other P-gp inhibitors are thought to be verapamil, cyclosporine A, reserpine, yohimbine and tamoxifen.96 Attempts are being made to develop highly selective P-gp inhibitors that will not inhibit CYP3A4 or other transporters.96
Similarly, rifampicin and St John's wort appear to be inducers, as demonstrated by the effects of some drugs such as digoxin87,95 and fexofenadine,95 which are P-gp substrates but are not metabolized by CYP3A4. The complexity of studying P-gp inducers is suggested by the fact that rifampicin may initially be a P-gp inhibitor and increase digoxin absorption, but after some time (one week) it may be a P-gp inducer and decrease its absorption.94
The best proof that the science on transporters in general and on P-gp in particular may not be ready for clinical practice is that a review of drug prescribing information from the USA and Europe indicates that frequently information on the same drug does not agree concerning the role of the transporter or even the terminology.87
Drug interactions in neuropsychopharmacologyThe P-gp articles on neuropsychopharmacology provide a very confusing picture for clinicians. Akamine et al.97 describe almost all antidepressants and antipsychotics as P-gp substrates and report that all of them can be inhibitors. On the other hand, Moons et al.93 suggest that not all second-generation antipsychotics may be P-gp substrates and O’Brien et al.98 acknowledge that it is not well established whether P-gp inhibition by antidepressants is clinically relevant or not. The confusing information becomes clearer after reading an AED review, where Zhang et al.99 state that (1) different in vitro models of P-gp function provide different results concerning whether AEDs are substrates or not, and (2) there is no agreed criteria to define P-gp substrates among AEDs. Articles tend to describe potent inducers such as carbamazepine and phenytoin as P-gp inducers. Valproic acid usually is described not as a P-gp substrate but as a P-gp inducer.97 The limited information available suggests that benzodiazepines may not be P-gp substrates,100,101 but may be substrates to other efflux transporters located in the BBB.102
To further acknowledge our current limitations in understanding DDI mediated by P-gp, it should be mentioned that Lin103 proposed that animal studies suggest that DDI at the transport level may have as much impact on tissue distribution, particularly in the brain, as on the systemic exposure measured by plasma concentrations. If that is true, it will complicate DDI interpretation for clinicians since the first step for interpreting a DDI is to measure plasma concentrations for increases or decreases. One should assume a pharmacokinetic component, but if not, one could then consider a pharmacodynamic component. If Lin103 is correct that P-gp DDIs have the potential to be pharmacokinetic DDIs, that does not influence TDM; then, these P-gp DDIs may be confused with pharmacodynamic DDIs.
Induction mechanism and nuclear receptorsThe induction of metabolic enzymes, such as CYPs and UGTs, and transporters, such as P-gp, implies that the amount of these proteins increases when they are induced. An increased quantity can be reached by increasing synthesis or by decreasing degradation. The induction of most metabolic enzymes and transporters appears to be mainly mediated by increased synthesis. There are two known exceptions: CYP2E1 and CYP2D6. CYP2E1 induction appears to be mediated by a completely different mechanism at the post-transcriptional level, leading to a stabilization of the protein that delays its destruction. CYP2E1 has a short half-life. CYP2E1 inducers such as ethanol and isoniazid increase CYP2E1 half-life by decreasing its degradation. CYP2D6 has always been considered a CYP isoform that cannot be induced; therefore, it is surprising that CYP2D6 activity increases during pregnancy. In a recent study using CYP2D6-humanized mice, Koh et al.24 have proposed that during pregnancy there is a decrease of a CYP2D6 repressor.
Increased synthesis is frequently mediated by a group of receptors that activate genes in the nucleus of the cell and are usually called nuclear receptors. As nuclear receptors have the ability to directly bind to DNA and regulate the expression of adjacent genes, they are considered transcription factors.
Nuclear receptors consist of 3 major protein domains: (1) a highly conserved DNA-binding domain which links the receptor to specific promoter regions of their target genes, (2) a less conserved ligand-binding domain, and (3) an area that recognizes other transcription factors and coactivators.104 There is major cross-talk between different nuclear receptors and other transcription factors.105 As a matter of fact, one of the receptors described in this section, the aryl hydrocarbon receptor (AhR), is not a nuclear receptor but it is part of another superfamily of transcription factors.
The most important nuclear receptors involved in induction include the pregnane X receptor (PXR) and the constitutive androstane receptor (CAR). It is believed that the glucocorticoid receptor (GR)106 and the estrogen receptors (ERs)107 may be involved in some induction phenomena. The estrogen receptors may be particularly important in the induction of some enzymes during pregnancy.107
Some nuclear receptor families include the receptors of steroid (including GR and ER) and thyroid hormones that are located in the cytosol but are moved to the nucleus after binding to the substrate. There are other receptors that were initially called orphan receptors since their function was not known.108 PXR and CAR are included in this category of orphan nuclear receptors; their respective official classifications are NR1I2 and NR1I3.109,110
One of the problems for clinicians trying to become familiar with this research area is its complexity. The role of nuclear receptors goes beyond the effects of xenobiotics such as drugs, pesticides, environmental pollutants, carcinogens, or some complex nutrients (e.g., flavonoids), or toxicology and pharmacology. Nuclear receptors call into play basic physiology since endogenous compounds including biliary compounds, hormones and vitamins bind to these nuclear receptors. As a matter of fact, these receptors regulate one of the most complex biosynthesis processes, the transformation of cholesterol into multiple complex biological agents with a complex molecular structure derivate from cholesterol including bile acids, corticoids, sexual hormones and vitamin D. To further complicate the literature, there are some important differences between human and rodent nuclear receptors, making the literature rather complex. As the literature in these nuclear receptors is in its early phases of development and is extraordinarily complex, it frequently provides contradictory information. In summary, this section provides some hints about how AED inducers may influence CYPs, but no attempt is made to summarize the mechanisms of UGT or P-gp induction. Although they are probably driven by the same nuclear receptors, the published information on UGT or P-gp is so contradictory104,111,112 that it is not possible to provide a coherent summary for clinicians.
AhRAhR is reviewed first in this section since it is not really a nuclear receptor. In the absence of a ligand, AhR is located in the cytosol but after binding it gets into the cell nucleus and becomes a transcription factor. AhRs are widely distributed in many tissues and are probably important for embryological development. Several inducers, including some drugs such as omeprazole, cruciferous vegetables and polycyclic aromatic hydrocarbons (PAHs) present in smoke, roasted coffee and charbroiled meat, bind to AhR, which is important for the induction of the CYP1 family, particularly CYP1A2 and some UGTs.
PXRPXRs are mainly located in the liver and small intestine. Carbamazepine, phenytoin, phenobarbital, rifampin and St. John's wort activate PXRs. PXRs are important for the induction of CYP2 and CYP3 families and some UGTs. In an in vitro study, Sinz et al.113 explored compounds that activate PXRs and classified them according to their potency as CYP3A4 inducers in three groups (rifampicin was in the first group of more active compounds; phenytoin and phenobarbital in the second group and carbamazepine in the third group).
CARCAR is only present in mammals and is mainly located in the liver and kidney. Phenytoin and phenobarbital are believed to activate CAR but the process is rather complex and not well understood. CAR is an important inducer of CYP2 and CYP3 families and UGT1A1. According to Pascussi et al.,105 CAR exhibits some pronounced selectivity for CYP2B6 when compared with CYP3A4, but PXR regulates both without selectivity.
ERAccording to in vitro studies (1) the high levels of estrogens (multiplied by 100) during pregnancy activate both ER and CAR, which has synergistic effects increasing CYP2B6 expression,107 and (2) ER may be partly responsible for the UGT1A4 induction seen with high estrogen levels, which may explain lamotrigine induction during pregnancy.114
GRCYP3A4 induction by corticoids such as dexamethasone, prednisolone and methylprednisolone is partly mediated by GR, although PXR may also be important.115
Genetic variationsThere are published attempts to initially explore how genetic variations at AhR, PXR and/or CAR can explain differences between individuals in response to inductions.109 The difficulty with interpreting these studies is that all review articles in the area insist that nuclear receptors and AhR appear to work in a coordinated way with great overlap among these transcription factors and with broad actions at CYPs, UGTs, and transporters. Therefore, to study how their genetic variations influence the induction of a drug that, for example, is mainly metabolized by CYP1A2, one may need to study all the genetic variations at the same time (CYP1A2, AhR, PXR and CAR).
Complex pharmacokinetic polypharmacy may require modifying correction factorsThis article provides correction factors in Table 3 with the idea of helping to correct for inductive effects in neuropsychopharmacology, which then allows modifying drug dosages to approximate for the effect of inducers in a specific patient. The literature, however, warns that extrapolation to an average subject is problematic since there is high variability in the population of the effects of inducers.116 Unfortunately, it is accurate to describe correction factors as gross approximations, but it is better to have them than not to have them.
This fifth section reviews four situations that may modify the inductive effects: non-oral route, PM individuals, presence of inhibitors and presence of other inducers.
Non-oral routes, particularly the IV route, are much less influenced by induction than the oral route.117 Subjects who do not have an active isoenzyme, such as PMs for CYP2D6 or CYP2C19, cannot be induced in that missing CYP but they can be induced in other CYPs. Risperidone is mainly metabolized by CYP2D6, but its metabolism is impaired in CYP2D6 PMs, who have approximately half the capacity to metabolize risperidone118 and need approximately half the average dose.119 CYP inducers cannot induce CYP2D6 but can induce CYP3A4, which is an auxiliary metabolic enzyme of risperidone metabolism. As a matter of fact, in the average risperidone subject taking potent CYP3A4 inducers, CYP3A4 probably becomes the most important metabolic enzyme for risperidone metabolism; this subject needs twice the risperidone dose.119 This approximated correction factor indicates that taking a potent CYP3A4 inducer (correction factor 2) and being a CYP2D6 PM (correction factor 0.5) cancel each other out, so these subjects have approximately a normal clearance. In the real world, CYP2D6 PMs taking potent CYP3A4 inducers have a slightly lower risperidone metabolism than normal.119
It is difficult to predict the outcomes in situations of combinations of inducers and inhibitors. In the case of risperidone, CYP3A4 inducers have more powerful effects that a potent risperidone inhibitor such as fluoxetine which inhibits both risperidone metabolic enzymes.119 In this situation of combining inhibitors and inducers, it is better to use TDM to establish the outcome. Table 1’s level of induction measured by “+” can help clinicians. The effects of potent inducers on CYP3A4 and CYP2B6 are massive (4+s in Table 1) and much more important than inhibitor effects. CYP1A2 induction is probably only moderate (2+s in Table 5) and, according to our experience with clozapine potent inducers, tends to have less potency than potent inhibitors such as fluvoxamine.120 On the other hand, CYP2C9 and CYP2C19 induction is minimal (1+ in Table 5) and clearly lower than the effect of potent inhibitors. Phenytoin is metabolized by CYP2C9 and CYP2C19 and is an inducer of its own metabolism but these inductive effects are probably not very clinically relevant. Phenytoin in high concentrations is a potent inhibitor of CYP2C9 and CYP2C19, which is quite clinically relevant during phenytoin intoxication. In the case of drugs metabolized by UGTs, such as lamotrigine, potent AED inducers require duplicating the dose, whereas valproate, a potent inhibitor, requires cutting the dose in half. The combination of potent inducers such as phenytoin and valproate require using normal lamotrigine doses. In this situation it may be wiser to use lamotrigine TDM since valproate inhibitory effects may be stronger.74
The literature provides very little recommendations on how to categorize what happens with combinations of inducers. The author has experience with two types of situations using combinations of (1) potent AED inducers with other inducers of CYP1A2, and (2) potent and mild AED inducers of CYP3A4 drugs. When patients are taking drugs metabolized by CYP1A2, smoking appears to influence induction by AhR, whereas induction by AEDs is mediated by PXR and/or CAR. Therefore, smoking and AED inducers appear to have additive and independent effects.120
When patients are taking CYP3A4 drugs, the effect of potent inducers such as carbamazepine, phenytoin or phenobarbital is much more potent than those of mild CYP3A4 inducers such as clobazam, eslicarbazepine, oxcarbazepine, rufinamide or topiramate (Table 4). In those cases, adding a mild inducer when a patient is taking a potent inducer may have no effect. Conversely, switching from a potent inducer such as carbamazepine to oxcarbazepine is associated with a decrease in inductive effects (increase in CYP3A4 substrate levels) and switching from a mild inducer such as oxcarbazepine to carbamazepine is associated with an important increase in inductive effects (decrease in CYP3A4 substrate levels). There are not many comparisons in the literature of CYP3A4 inducers, but Ohno et al.,121 after reviewing the literature using a complex mathematical model, provide some comparisons of the effects of various CYP3A4 inducers. They described metabolism increases of 7.7 by rifampin (450–600mg/d); 4.7 by phenytoin (300–400mg/day); 3.0 by carbamazepine (200–600mg/day); 1.4 by efavirenz, an antiretroviral agent (600mg/day); and 1.2 by St. John's wort (600–900mg/day).
Pharmacodynamic DI may also contribute to modification of inducer effectsThe correction factors (Table 3) help clinicians to interpret the effect of adding potent inducers to one drug. The prior section tries to give clinicians a first taste of complexity by explaining that the route, the individual with PM status, or the co-prescription of inhibitors or other inducers must also be taken into account when interpreting AED inducer effects. This section tries to focus on the further complexity of real-world prescription by describing, as examples, combinations in pairs of three important AEDs, carbamazepine, phenytoin and valproic acid; carbamazepine-valproic acid, carbamazepine-phenytoin and phenytoin-valproic acid. The author has seen hundreds of epileptic and/or psychiatric patients taking these combinations and has found that usually the neurologists and/or psychiatrists managing these patients have no clue about the complexity of these DDIs, probably the most complicated DDIs in neuropsychopharmacology, and the most uncertain in their outcome.
Part I describes (1) phenytoin as a powerful inducer of multiple enzymes, which in high plasma concentrations can saturate CYP2C9 and CYP2C9, and (2) many of the drugs called mild inducers in this article, including VPA, as drugs that can be clinically relevant inhibitors, too. Thus, a first level of complexity is that drugs frequently can be both inducers and inhibitors. Table 8 tries to provide a taste of how difficult it can be to interpret these DDIs, since they may combine both complex pharmacodynamic and pharmacokinetic components. To master these three AED DDIs, one must master other levels of complexity by knowing that protein binding is very important for phenytoin and VPA, and possibly relevant at times for carbamazepine. Another level of complexity is that pharmacodynamic mechanisms are also important in interpreting DDIs. Since this review article focuses on induction, it cannot review protein binding or pharmacodynamics in detail, but Table 8 provides a comprehensive description of complex pharmacological mechanisms of three DDIs. Other prior publications provide more comprehensive information, including pharmacodynamic mechanisms and protein binding.74,122,123 In summary, the author proposes that psychiatrists and neurologists should not use these three combinations unless they thoroughly understand the pharmacological mechanisms behind DDIs and use TDM.
Complex pharmacokinetic and pharmacodynamic AED combinations.
| Carbamazepine and VPA |
| Summary for clinicians: be very careful and use TDM, including free concentrations |
| Pharmacokinetics effects |
| Effects of VPA on serum carbamazepine concentrations: ↑ total and free |
| (−) epoxide hydoxylase: ↑ serum epoxide concentration |
| (−) UGT2B7: ↓ serum glucuronidate diol metabolite |
| Displace carbamazepine from binding to plasmatic proteins |
| Effects of carbamazepine on serum VPA concentrations: ↓ total and ↑ free |
| Probably inhibition of some metabolic enzymes |
| Displace VPA from binding to plasmatic proteins |
| Pharmacodynamics effects (poorly understood): |
| Carbamazepine blocks of voltage-gated sodium channels and VPA have complex anti-convulsant effects |
| Possible additive mood stabilizer effects by acting at the intracellular signaling system |
| Textbooks usually report increased risk for neurological ADRs |
| Carbamazepine and phenytoin |
| Summary for clinicians: Be very careful and use TDM, including free concentrations |
| Pharmacokinetics |
| Effects of phenytoin on serum carbamazepine concentrations: ↓ total and ↑ free |
| Further enzymatic induction |
| Displace carbamazepine from binding to plasmatic proteins |
| Effects of carbamazepine on serum phenytoin concentrations: unclear |
| Possible further enzymatic induction |
| Displace phenytoin from binding to plasmatic proteins |
| Possible CYP2C19 inhibition |
| Pharmacodynamics (poorly understood): |
| Probably additive anti-convulsant and ADRs: both are blockers of voltage-gated sodium channels |
| Phenytoin and VPA |
| Summary for clinicians: Be very careful and use TDM, including free concentrations. |
| Pharmacokinetics |
| Effects of phenytoin on serum VPA concentrations: ↓ total and ↑free |
| Enzymatic induction |
| Displace VPA from binding to plasmatic proteins |
| Effects of VPA on serum phenytoin concentrations: ↑ total and free |
| Powerful CYP2C9 inhibition |
| Displace phenytoin from binding to plasmatic proteins |
| Pharmacodynamics (poorly understood): |
| Phenytoin blocks of voltage-gated sodium channels and VPA have complex anti-convulsant effects |
(−): inhibition; ADRs: adverse drug reactions; AED: antiepileptic drugs; TDM: therapeutic drug monitoring; VPA: valproic acid.
During the last 15 years as a clinician and consultant for difficult patients in neuropsychopharmacology, the author has found a small number of patients with extremely unusual pharmacokinetic profiles who appear to be extremely sensitive to inductive effects and need massive doses of some drugs to get therapeutic serum concentrations. After many years of consideration, the author thinks that the best way to understand them would be to say that they are candidates for unusual genetic profiles at the nuclear receptor levels, which makes them extremely sensitive to inductive effects across different metabolic pathways.
Table 9124–128 lists six patients the author identified as possibly having an excessive response to inducers and thus followed. The prevalence of these unusual individuals is unknown, but the author estimates they make up between 1/1000 and <1/100 of patients with severe mental illness. Two of the six patients needed very high doses of CYP3A4 substrates in the presence of AED potent inducers (Cases 1 and 3). One patient appeared to be very sensitive to UGT induction; he needed extremely high doses of lamotrigine and lorazepam (Case 5). Four of the six patients appeared to have major inductive effects secondary to valproic acid demonstrated by TDM effects on valproic acid (Cases 1 and 4), clozapine (Case 2) and olanzapine (Case 3). The most perplexing valproic acid case was a case of a patient who developed a metastatic renal carcinoma in the context of tuberous sclerosis and required >10g/day of divalproex sodium to get therapeutic serum concentrations of valproic acid when co-medicated with phenytoin (Case 6). Three UGTs, UGT1A6, UGT1A9, and UGT2B7, are present in high concentration in the kidneys.71 As described in Part I, UGT1A6, UGT1A9, and UGT2B7, are probably involved in the valproic acid metabolism.74
List of patients with extreme sensitivity to inductive effects.
| Case 1: Caucasian ♂ inpatient with schizophrenia and late-onset epilepsya |
| (A) Extreme sensitivity to CYP3A4 induction |
| -Carbamazepine: 1500–2000mg/dayb was needed in the past to reach therapeutic serum concentrations |
| -Quetiapine: |
| On phenytoin and VPA, the quetiapine C/D ratio was 10 times lower than expected |
| On VPA, the quetiapine C/D ratio was 2–6 times lower than expected |
| -Diazepam:c On phenytoin, serum concentrations were undetected despite taking 30mg/day |
| On VPA, diazepam clearance after 30mg IM was ≥3 times higher than expected |
| (B) Possible VPA auto-induction when taking concentrate, but not present on divalproex sodium |
| On concentrate, 5250mg/dayd was needed to reach therapeutic concentrations |
| On divalproex sodium, 2000mg/daye was enough for therapeutic concentrations |
| (C) Metabolism of clozapine and olanzapine (CYP1A2 drugs) was normal for a male smokerf |
| Case 2: African-American ♂ inpatient with schizoaffective disorderg |
| (A) Extreme clozapine induction by VPA (divalproex sodium) |
| -Smoking (20 cigarettes/day): ≥ 650mg/day was needed for reaching serum concentrations >350ng/mlh |
| During smoking induction, the patient demonstrated a high metabolic capacity for clozapine |
| -Smoking and VPA: ∼1200mg/day was needed for reaching serum concentrations >350ng/mli |
| Addition of VPA induction led to the highest clozapine metabolic capacity the author has seen |
| Case 3: Caucasian ♂ inpatient with schizoaffective disorderj |
| (A) Extreme sensitivity to CYP3A4 induction |
| -Carbamazepine: Up to 2800mg/dayb was needed to reach therapeutic serum concentrations; this is partly explained by deposits in fat tissue and high volume of distributionk |
| -Risperidone: On carbamazepine, approximately 4 times higher than normal metabolic capacity |
| On divalproex sodium, risperidone metabolism was normal |
| -Paliperidone: On carbamazepine, high capacity to metabolize paliperidone |
| (B) Extreme olanzapine induction by VPA (divalproex sodium) and omeprazole |
| -Olanzapine: On divalproex sodium and omeprazole, 1.5–2 times higher than normal metabolism |
| Case 4: Caucasian ♂ inpatient with bipolar disorderl |
| A)Possible VPA auto-induction when taking divalproex sodium |
| -Discharged on 4000mg/daym |
| (A) Extreme sensitivity to UGT induction during phenytoin and phenobarbital treatment |
| -Lamotrigine: 2.6 times higher than the recommended dosage was neededo |
| -Lorazepam: Doses >20mg/day were tolerated with no sedation |
| Case 6: Caucasian ♂ inpatient with tuberous sclerosis and kidney tumorsp |
| (A) Increased VPA metabolic capacity when taking phenytoin and VPA |
| -Divalproex sodium: Dose increased >10,000mg/day to reach therapeutic serum concentrationsq |
| -Phenytoin: No changes in phenytoin metabolism and no need for changes in dosage |
Usual carbamazepine doses to reach therapeutic concentrations: 800–1200mg/d. Maximum recommended dose is 1600mg/day.124
Diazepam is metabolized primarily by CYP2C19; CYP3A4 is an auxiliary enzyme. In this patient CYP3A4 was probably the primary metabolic enzyme for diazepam.
At the time of highest carbamazepine dose, BMI was 40 with weight of 191kg. The high dose is partly explained by obesity.128
Patient needed 1600mg/day to get therapeutic serum lamotrigine concentrations. The maximum recommended dose is 600mg/day.
Followed for 4 years (ages 44 to 48 until he died). He initially had angiomyolipomas in both kidneys. In the second year, a growing right kidney mass led to possible diagnosis of renal carcinoma and nephrectomy pathology that suggested angiomyolipoma. In the third year, brain metastasis became evident.
In the beginning when the patient had bilateral kidney tumors, he needed around 5000mg/day of divalproex sodium to get therapeutic concentrations with VPA C/D ratios of 0.010–0.018. After nephrectomy and obvious metastatic renal cancer was present, he needed 10,500mg/day of divalproex sodium to get therapeutic concentrations with VPA C/D ratios of 0.005–0.009.
This two-part article provides information for clinicians to compensate for the oversight in the literature on AED inducers. Part I introduces the subject of potent (Table 1) and mild inducers (Table 4) and provides clinical recommendations on correcting the effect of inducers through dose modifications of the induced substrates by using correction factors (Table 3).
Part II attempts to educate clinicians about the complex nature of interpreting AED DDIs by providing basic pharmacological knowledge to help improve their ability to interpret complex DDIs.
The basic pharmacology of CYPs, UGTs, P-gp, and nuclear receptors is reviewed. Six CYP isoenzymes, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, are definitively important for the metabolism of neuropsychopharmacological drugs. CYP2B6 and CYP3A4 are very sensitive to induction. CYP1A2 is moderately sensitive while CYP2C9 and CYP2C19 are only mildly sensitive. Finally, CYP2D6 cannot be induced by medications (Table 5). The genetic variations of CYP2C9, CYP2C19 and CYP2D6 are understandable enough for clinical use. PMs are subjects who lack specific active CYPs; while they have been identified for CYP2C19 and CYP2D6, their frequencies vary with race (Table 6).
UGTs are the most important of the conjugation enzymes (traditionally called Phase II metabolic enzymes). They are the main metabolic enzymes for a few antipsychotics, a few AEDs, and a few benzodiazepines (Table 7). The literature has neglected UGTs; they are less well understood than CYPs.60 Several important UGTs in neuropsychopharmacology include UGT1A1, UGT1A3, UGT1A4, UGT2B7 and UGT2B15 (Table 7). There is no definitive information on how inducers influence their activity. Similarly, it is not clear how genetic variations or inhibitors (such as valproic acid) influence any of these specific UGTs.
Another major group of pharmacokinetic proteins called transporters can also be induced. They usually work in tandem with metabolic enzymes to eliminate xenobiotics from the body. P-gp is the most important transporter and is a major component of the BBB. P-gp science may not be ready for clinical practice. US prescribed information and European prescribed information disagree on the relevance of transporters and the terminology used to describe them. Moreover, the literature does not agree on which neuropsychopharmacological drugs are substrates, inhibitors or inducers for P-gp.
The new and rapidly expanding literature on nuclear receptors is beginning to provide some understanding of how inducers work. The induction of metabolic enzymes such as CYPs and UGTs, and transporters such as P-gp, implies that the amount of these proteins increases during induction. This increase is almost always explained by increased synthesis mediated by a group of receptors that activate genes in the nucleus of the cell, usually called nuclear receptors. Nuclear receptors are considered transcription factors because they can directly bind to DNA and regulate the expression of adjacent genes. The most important nuclear receptors involved in induction are PXR and CAR. The hormonal receptors GR and ER may also have some role in induction. AhR is not a nuclear receptor but is part of another superfamily of transcription factors and mediates smoking's inductive effects on CYP1A2. The literature has initiated exploring how genetic variations at AhR, PXR and/or CAR may explain differences between individuals in response to inductions. Nuclear receptors and AhR appear to work in a coordinated way with great overlap among these transcription factors and with broad actions at CYPs, UGTs, and transporters. Therefore, establishing the induction role of specific genetic variations at individual genes will not be easy.
Table 3 provides correction factors for AED inducers. However, the literature warns that extrapolation to an average subject is problematic since there is high variability in the population of the effects of inducers. Non-oral routes, particularly the IV route, are much less influenced by induction than the oral route. PMs cannot be induced by the CYP as they are missing, but they can be induced in other CYPs. It is difficult to predict outcomes when combinations of inducers and inhibitors are present. The literature provides very few recommendations on how to explain what happens with these combinations of inducers. The author has experience with two types of situations using combinations of (1) potent AED inducers with other inducers of CYP1A2, and (2) potent and mild AED inducers of CYP3A4 drugs. When patients are taking drugs metabolized by CYP1A2, smoking appears to influence induction by AhR, whereas induction by AEDs is mediated by PXR and/or CAR. Therefore, smoking and AED inducers appear to have additive and independent effects. When patients are taking CYP3A4 drugs, the effect of potent inducers such as carbamazepine, phenytoin or phenobarbital is much more potent than those of mild CYP3A4 inducers such as clobazam, eslicarbazepine, oxcarbazepine, rufinamide or topiramate (Table 4). In those cases, adding a mild inducer when a patient is taking a potent inducer may have no effect.
Table 8 uses as an example the combinations of carbamazepine-valproic acid, carbamazepine-phenytoin and phenytoin-valproic acid to demonstrate the complexity of understanding some AED DDIs. To understand the outcome of these three pairs of combinations, it is not only necessary to take into account inductive and inhibitory effects, but pharmacodynamic DDIs and protein binding. Other prior publications provide more comprehensive information, including pharmacodynamic mechanisms and protein binding, for understanding DDI in neuropsychopharmacology.74,123,124
During his last 15 years as a clinician and consultant for difficult patients in neuropsychopharmacology, the author has found a small number of patients who have extremely unusual pharmacokinetic profiles, probably due to unusual genetic profiles at the nuclear receptor level, which makes them extremely sensitive to AED inductive effects across different metabolic pathways. Table 9 describes six patients including: (1) two needing very high doses of CYP3A4 substrates in the presence of AED potent inducers; (2) four with major inductive effects secondary to valproic acid; (3) one very sensitive to UGT induction, requiring extremely high doses of lamotrigine and lorazepam; and (4) a patient with tuberous sclerosis who needed to take >10g/day of divalproex sodium to get therapeutic serum concentrations of valproic acid when taken in combination with phenytoin.
Conflict of interestNo commercial organizations had any role in the writing of this paper for publication. The author reports no financial relationship with commercial interests in the last 36 months.
The author acknowledges the work of Lorraine Maw, MA, and Margaret T. Boden, RN, MLT, the Center for Health Research Mental at Eastern State Hospital, Lexington, KY, United States, for their collaboration for editing this article.
Please cite this article as: de Leon J. Efectos de los inductores antiepilépticos en la neuropsicofarmacología: una cuestión ignorada. Parte II: cuestiones farmacológicas y comprensión adicional. Rev Psiquiatr Salud Ment (Barc.). 2015;8:167–188.
Tables 1–4 were developed for part I of this article but are included here to facilitate the lecture.