



Es reconocido que los factores genéticos están involucrados en la etiología de diversas epilepsias, sin embargo los genes causales se han logrado identificar principalmente en las epilepsias monogénicas, que representan sólo el 1 a 2% de los síndromes epilépticos. El presente artículo describe algunos de los principales genes identificados hasta el momento en los síndromes mendelianos y no mendelianos. También se hace mención de los principales genes involucrados en la etiología de las malformaciones del desarrollo cortical y de las epilepsias mioclónicas progresivas.
Las epilepsias forman un grupo de síndromes neurológicos crónicos derivados de alteraciones de las funciones cerebrales, asociadas o no a otras condiciones patológicas. Los síndromes epilépticos se clasifican en sintomáticos, criptogénicos e idiopáticos (1). En relación a las epilepsias sintomáticas, las crisis epilépticas representan un síntoma de una lesión estructural en el sistema nervioso; las criptogénicas presentarían una presumible base orgánica, pero sin etiología definida (1,2). Las epilepsias idiopáticas son aquellas sin sustrato lesional conocido, probablemente relacionadas a alguna predisposición genética; idiopático significa que la propia epilepsia es la enfermedad y no un síntoma de alguna otra condición (1,2).
En las décadas de los 50 y 60, algunos estudios epidemiológicos mostraron las primeras evidencias científicas de predisposición genética en diferentes síndromes epilépticos (3, 4). Estudios más recientes en gemelos confirmarían el importante impacto de los factores genéticos en la etiología de las epilepsias (5).
A pesar de reconocer que los factores genéticos están involucrados en la epilepsias, la identificación de los genes que causan o predisponen a la enfermedad ha sido dificultoso, puesto que las epilepsias, particularmente las idiopáticas, son enfermedades complejas. Las enfermedades complejas son definidas como condiciones en que la correspondencia entre genotipo y fenotipo no es completa (6).
Los mayores problemas asociados con el estudio de enfermedades complejas son: Penetrancia incompleta (presencia del alelo que predispone a la enfermedad, pero sin manifestación clínica); Heterogeneidad genética (mutaciones en diferentes genes que resultan en un mismo fenotipo); Herencia poligénica (la manifestación de la enfermedad necesita de la presencia de mutaciones en múltiples genes) o multifactorial (factores genéticos y ambientales influenciando la manifestación clínica de la enfermedad) y; la alta prevalencia en la población (6, 7).
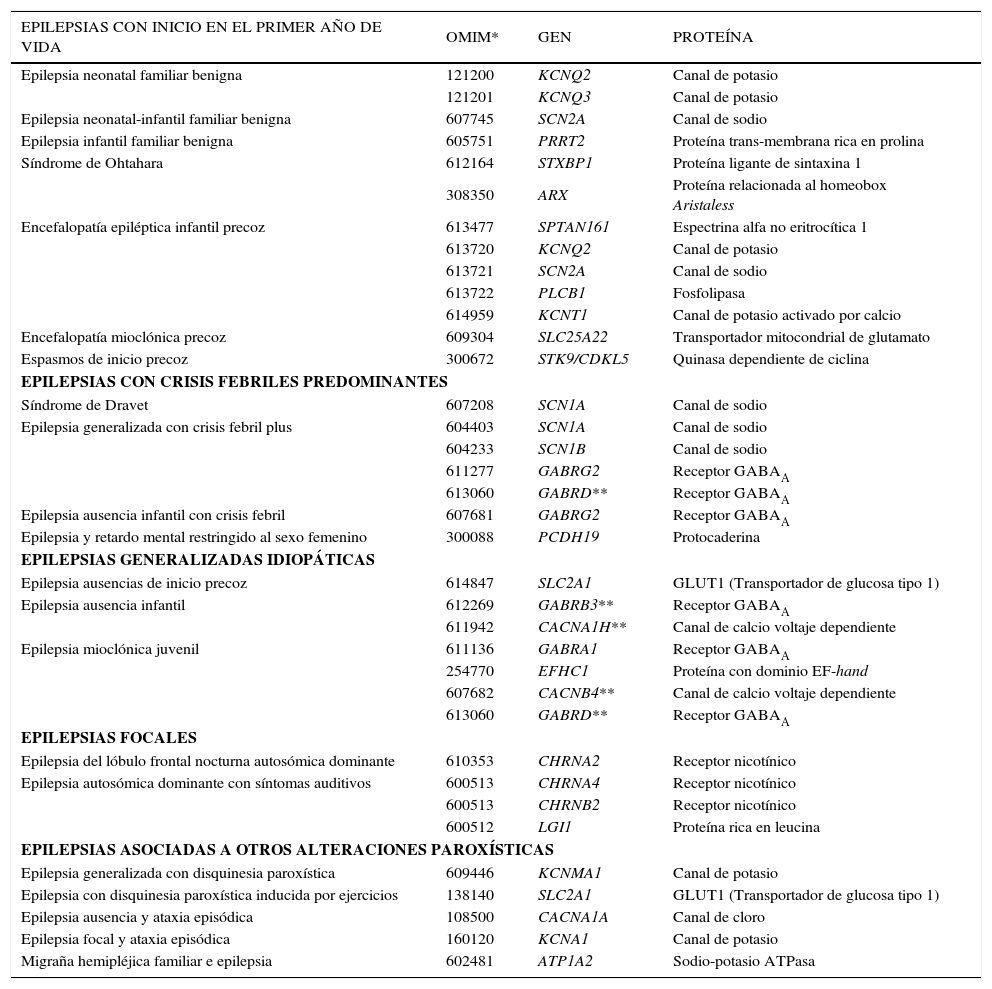
Genes y epilepsiasUna mutación en una de las sub-unidades de un receptor colinérgico (CHRNA4) fue la primera mutación descrita relacionada con una epilepsia idiopática. Esta se encontró en una familia con epilepsia frontal nocturna autosómica dominante en 1995 (8). En las epilepsias generalizadas, las primeras mutaciones descritas fueron en familias con epilepsia neonatal familiar benigna, en los genes KCNQ2 y KCNQ3, los cuales codifican secuencias de las subunidades de canales de potasio voltaje dependiente (9,10).
En las últimas décadas, se ha identificado un número creciente de mutaciones asociadas a epilepsias, principalmente en síndromes epilépticos monogénicos raros; sin embargo, apenas el 1 a 2% de las epilepsias idiopáticas parecieran ser monogénicas (11). Ejemplos de síndromes monogénicos o mendelianos son las epilepsias del lóbulo frontal nocturna autosómica dominante y las epilepsias mioclónicas progresivas, en las cuales mutaciones de un único gen son suficientes para producir crisis epilépticas.
Los principales genes relacionados a epilepsias hasta este momento se muestran en la Tabla 1. La mayoría de ellos codifica proteínas de sub-unidades de canales iónicos y una gran parte participa en la generación de otros proteínas que integran esos canales (11). Esta relación resulta bastante plausible, dado que estos canales iónicos forman parte de los procesos de excitabilidad neuronal y, por lo tanto, alteraciones de estas proteínas pueden perturbar el equilibrio en la comunicación entre neuronas, lo que podría resultar en descargas epilépticas. También se reconoce la existencia de genes involucrados en otras vías metabólicas asociadas a epileptogénesis, que si bien pueden tener funciones importantes, aun han sido poco explorados.
Principales genes relacionados a epilepslas hasta este momento
| EPILEPSIAS CON INICIO EN EL PRIMER AÑO DE VIDA | OMIM* | GEN | PROTEÍNA |
|---|---|---|---|
| Epilepsia neonatal familiar benigna | 121200 | KCNQ2 | Canal de potasio |
| 121201 | KCNQ3 | Canal de potasio | |
| Epilepsia neonatal-infantil familiar benigna | 607745 | SCN2A | Canal de sodio |
| Epilepsia infantil familiar benigna | 605751 | PRRT2 | Proteína trans-membrana rica en prolina |
| Síndrome de Ohtahara | 612164 | STXBP1 | Proteína ligante de sintaxina 1 |
| 308350 | ARX | Proteína relacionada al homeobox Aristaless | |
| Encefalopatía epiléptica infantil precoz | 613477 | SPTAN161 | Espectrina alfa no eritrocítica 1 |
| 613720 | KCNQ2 | Canal de potasio | |
| 613721 | SCN2A | Canal de sodio | |
| 613722 | PLCB1 | Fosfolipasa | |
| 614959 | KCNT1 | Canal de potasio activado por calcio | |
| Encefalopatía mioclónica precoz | 609304 | SLC25A22 | Transportador mitocondrial de glutamato |
| Espasmos de inicio precoz | 300672 | STK9/CDKL5 | Quinasa dependiente de ciclina |
| EPILEPSIAS CON CRISIS FEBRILES PREDOMINANTES | |||
| Síndrome de Dravet | 607208 | SCN1A | Canal de sodio |
| Epilepsia generalizada con crisis febril plus | 604403 | SCN1A | Canal de sodio |
| 604233 | SCN1B | Canal de sodio | |
| 611277 | GABRG2 | Receptor GABAA | |
| 613060 | GABRD** | Receptor GABAA | |
| Epilepsia ausencia infantil con crisis febril | 607681 | GABRG2 | Receptor GABAA |
| Epilepsia y retardo mental restringido al sexo femenino | 300088 | PCDH19 | Protocaderina |
| EPILEPSIAS GENERALIZADAS IDIOPÁTICAS | |||
| Epilepsia ausencias de inicio precoz | 614847 | SLC2A1 | GLUT1 (Transportador de glucosa tipo 1) |
| Epilepsia ausencia infantil | 612269 | GABRB3** | Receptor GABAA |
| 611942 | CACNA1H** | Canal de calcio voltaje dependiente | |
| Epilepsia mioclónica juvenil | 611136 | GABRA1 | Receptor GABAA |
| 254770 | EFHC1 | Proteína con dominio EF-hand | |
| 607682 | CACNB4** | Canal de calcio voltaje dependiente | |
| 613060 | GABRD** | Receptor GABAA | |
| EPILEPSIAS FOCALES | |||
| Epilepsia del lóbulo frontal nocturna autosómica dominante | 610353 | CHRNA2 | Receptor nicotínico |
| Epilepsia autosómica dominante con síntomas auditivos | 600513 | CHRNA4 | Receptor nicotínico |
| 600513 | CHRNB2 | Receptor nicotínico | |
| 600512 | LGI1 | Proteína rica en leucina | |
| EPILEPSIAS ASOCIADAS A OTROS ALTERACIONES PAROXÍSTICAS | |||
| Epilepsia generalizada con disquinesia paroxística | 609446 | KCNMA1 | Canal de potasio |
| Epilepsia con disquinesia paroxística inducida por ejercicios | 138140 | SLC2A1 | GLUT1 (Transportador de glucosa tipo 1) |
| Epilepsia ausencia y ataxia episódica | 108500 | CACNA1A | Canal de cloro |
| Epilepsia focal y ataxia episódica | 160120 | KCNA1 | Canal de potasio |
| Migraña hemipléjica familiar e epilepsia | 602481 | ATP1A2 | Sodio-potasio ATPasa |
Adaptado de Ottman et al., 2010
Existen otros genes potencialmente relacionados con la epilepsia descritos en la literatura y que no han sido incluidos en la Tabla 1, como el gen BRD2 en epilepsia mioclónica juvenil (EMJ) (12) y el gen ME2 en diferentes epilepsias generalizadas idiopáticas (EGIs) (13). Esos genes fueron descubiertos en estudios de ligamiento genética, seguido por análisis de asociación, sin embargo aun son necesarios estudios de las mutaciones causales para confirmar esos resultados.
A pesar que desde hace mucho tiempo se ha observado un componente genético en las EGIs, en muy pocos casos se ha determinado alguna etiología genética (Tabla 1). Las EGIs comprenden varios fenotipos de crisis comunes, incluyendo clásicamente: epilepsia ausencia de la niñez (EAI), epilepsia ausencia juvenil, EMJ y epilepsia con crisis tónico-clónicas generalizadas al despertar (1). En estas epilepsias, las características de los síndromes se sobreponen y, además de eso, diferentes EGIs pueden ocurrir en una misma familia, lo que dificulta los hallazgos genéticos. El complejo patrón de herencia de las EGIs sugiere una interacción de varios genes de susceptibilidad, de forma que polimorfismos en diferentes genes contribuirían de forma aditiva a la enfermedad (13). De este modo, aunque se han identificado muchos loci, pocos genes han sido descritos como causales de EGIs. Uno de esos genes es el GABRA1, que fue encontrado alterado en individuos afectados con EMJ en una familia franco-canadiense (14). Mientras tanto y después de diez años, sólo un grupo de investigadores encontró una mutación en ese gen en un niño con EAI (15), reforzando la hipótesis de que el fenotipo de las EGIs comparte una base genética común.
A diferencia de las EGIs, la epilepsia infantil familiar benigna, la neonatal familiar benigna y la neonatal-infantil familiar benigna son síndromes que difieren fenotípica y genéticamente, a pesar de su semejanza de nombres (Tabla 1). En al año 2012 fueron descritas mutaciones del gen PRRT2 en 14 (82%) de 17 familias con epilepsia infantil familiar benigna (16). Durante ese mismo año, más de 20 artículos fueron publicados sobre el gen PRRT2 en epilepsia infantil, destacando la importancia de ese gen en esas epilepsias (17).
Otro gen que tiene una gran importancia en la genética de las epilepsias es el gen SCN1A (OMIM #182389). Las mutaciones en este gen pueden causar un espectro de manifestaciones convulsivas que van desde el inicio precoz de crisis febriles aisladas a epilepsia generalizada con crisis febriles plus (GEFs+), lo que representa el fenotipo más grave. Los pacientes con convulsiones febriles aisladas, que generalmente se inician entre los 6 meses y los 4 años de vida, muestran una remisión espontánea alrededor de los 6 años, sin embargo, los pacientes con GEFs+ continúan teniendo crisis febriles y no febriles hasta el final de la vida. Alteraciones en ese mismo gen también han sido descritas en individuos con síndrome de Dravet o epilepsia mioclónica grave de la infancia, siendo el fenotipo más severo y grave asociado a mutaciones del gen SCN1A (Tabla 1). Se ha encontrado mutaciones en heterocigotos en el 70 a 80% de los casos de Dravet; de estas mutaciones el 95% fueron mutaciones de novo (mutaciones no encontradas en los padres del individuo afectado), lo que puede explicar por que hermanos o hijos de padres con Dravet pueden no necesariamente ser afectados (18).
Se ha identificado otros genes en síndromes epilépticos sintomáticos mendelianos, donde las crisis serían síntomas de desórdenes más ampliamente distribuidos en el sistema nervioso central. Dentro de estos se encuentran las malformaciones del desarrollo cortical y las epilepsias mioclónicas progresivas.
Malformaciones de desarrollo corticalLas malformaciones del desarrollo cortical (MDC) constituyen una de las principales causas de retraso mental y epilepsia. Cerca del 8% de los pacientes con epilepsia que son tratados en centros especializados son portadores de alguna forma de MDC, lo que corresponde a la primera causa de cirugía de la epilepsia en niños y a la segunda etiología mas frecuente de epilepsia refractaria en adultos, siendo sólo superado por las epilepsias del lóbulo temporal asociada a esclerosis hipocampal (19). Los avances en la comprensión de los mecanismos básicos de la formación de la corteza y las técnicas de resonancia magnética han demostrado que las MDC también pueden derivar de factores genéticos y no solamente de eventos prenatales (20).
La Heterotopía Nodular Periventricular (HNP), que corresponde a una malformación de las etapas de migración neuronal, es caracterizada por la presencia de neuronas heterotópicas próximas a la región ventricular, en una corteza aparentemente normal. Han sido identificados hasta el momento dos genes involucrados en la etiología de la HNP: el gen FLNA (Xq28), que es responsable de una forma de heterotopía periventricular bilateral con un patrón clásico de herencia dominante ligada al cromosoma X (21); el gen ARFGEF2 (20q13.13), que esta asociado a una forma de microcefalia autosómica recesiva, epilepsia y retraso del desarrollo cortical (22). Otro gen que esta involucrado en varios tipos de malformaciones corticales es el gen WDR62, cuya función no es totalmente conocida. Bilügvar et al., en el año 2010, identificaron mutaciones patológicas en este gen en pacientes con malformaciones tan diversas como la microlisencefalia, agiria, paquigiria, esquicencefalia y microcefalia (23).
De esta forma, se descartan (o desechan) las nociones previamente aceptadas en el sentido que una determinada malformación pertenecería exclusivamente a una fase del desarrollo cortical, y que los genes involucrados tendrían funciones limitadas solamente a una etapa de la embriología de la corteza cerebral (24).
Epilepsias mioclónicas progresivasLas epilepsias mioclónicas progresivas (EMP) se refieren a un grupo de enfermedades neurodegenerativas con heterogeneidad clínica y genética, generalmente con síntomas debilitantes y gravedad variable. Son enfermedades raras, frecuentemente familiares y caracterizadas por crisis mioclónicas, crisis tónico-clónicas generalizadas y deterioro neurológico progresivo, particularmente con demencia y ataxia (25).
Las etiologías más frecuentes son la enfermedad de Unverricht-Lundborg, la enfermedad de Lafora, la lipofuscinosis neuronal ceroídea (LCNs), las encefalomiopatías mitocondriales y las sialidosis (25).
Gran parte de las EMPs tienen herencia genética autosómica recesiva y, por lo tanto, la enfermedad ocurre con mayor frecuencia, aunque no exclusivamente en hijos de padres consanguíneos.
El gen responsable de la enfermedad de Unverricht-Lunborg (OMIM nº 254800) CSTB, codifica para la proteína cistatina B, que es una enzima que pertenece a la familia de los inhibidores de la cisteína proteasa. Esta enzima tiene la función de inhibir la degradación celular después de la liberación de enzimas lisosomales en el citoplasma.
En la enfermedad de Lafora (OMIM nº 254780) dos genes han sido identificados: el gen EPM2 y el gen NHLRC1. El primero en ser identificado fue el EPM2, que codifica la proteína tirosina fosfatasa laforina; se han encontrado mutaciones en ese gen en más del 80% de los pacientes con esta enfermedad. El gen NHLRC1 codifica la proteína malina que es una sub-unidad de la ubiquitinaligasa 3. Algunos datos sugieren que la proteína malina formaría un complejo funcional con la proteína laforina, promoviendo con ello la ubiquitinación de proteínas involucradas en el metabolismo del glicógeno, esta alteración resultaría en la formación de los cuerpos de Lafora (26).
Las LCNs representan un gran grupo de enfermedades de depósito lisosomal. Estas ocurren en la infancia, adolescencia o en la edad adulta y, en las cuales un lipopigmento autofluorescente se acumula dentro de los lisosomas. Actualmente, ya se han descrito diez formas de LCNs con diferente incidencia alrededor del mundo, así como innumerables variantes (25). Dada la complejidad del diagnóstico molecular, el estándar de confirmación diagnóstica de las diferentes formas de LCN son los hallazgos hitopatológicos, a partir de una biopsia de piel de la axila o de conjuntiva ocular (25).
Las encefalomiopatías mitocondriales constituyen un grupo heterogéneo de enfermedades neurodegenerativas asociadas a diferentes mutaciones del DNA mitocondrial (mtDNA). Uno de los fenotipos más frecuentemente asociados a EMP es el síndrome de MERRF (epilepsia mioclónica con fibras rotas rojas – OMIM nº545000). En el 80 a 90% de los casos es causada por la mutación puntual A8344G en el mtDNA; sin embargo, otras 14 mutaciones puntuales en el mtDNA se han asociado al síndrome de MERRF (25). El patrón de herencia es mitocondrial, por lo tanto vía materna. El grado de heteroplasmía (porcentaje de DNA mutante y normal) en los diferentes tejidos, será lo que determinará la variabilidad del fenotipo. Otra EMP que también comparte la herencia mitocondrial es el síndrome MELAS (encefalopatía mitocondrial, acidosis láctica y episodios tipo-Stroke – OMIM nº 540000), siendo la mutación más frecuente la mutación puntual de tipo sentido opuesto en el nucleótido 3243 en el mtDNA.
Las sialidosis también forman parte del grupo de enfermedades lisosomales y están asociadas una deficiencia primaria de la enzima sialidasa (neuroaminidasa) y, en algunas formas a la deficiencia de betagalactosidasa. Solamente la sialidosis tipo 1 (OMIM nº256550), cuyo inicio es en la adolescencia, se ha asociado a EMP (25).
Pruebas molecularesLas pruebas genéticas consisten en el uso de información genética, tanto para proporcionar diagnósticos más claros en personas con la enfermedad declarada o con sospecha de ella (prueba diagnóstica), como para predecir el posible riesgo de la enfermedad en personas con riesgo aumentado debido a una historia familiar positiva (prueba predictiva) (7). La identificación de un gran número de genes involucrados en la etiología de enfermedades humanas ha resultado en un marcado aumento del uso de estas pruebas en la práctica clínica. Actualmente, más de 2000 pruebas genéticas están disponibles para uso clínico (1), siendo la mayoría dirigidos a trastornos genéticos raros que siguen patrones de herencia mendeliana. Sin embargo, el significado clínico de las pruebas genéticass puede producir confusión, dado que los resultados no siempre son de interpretación evidente y, en algunos casos el significado de la variación genética es incierto (27).
Aunque son diversos lo genes que han sido identificados en diferentes síndromes epilépticos, relativamente pocos han tenido utilidad clínica como para ser utilizados como pruebas genéticas en la actualidad (7). Algunos ejemplos de pruebas genéticas con importantes implicancias clínicas son los usados en sospechas de epilepsias del lóbulo frontal nocturna autosómica dominante, epilepsia ausencia de la niñez de inicio precoz, síndrome de Dravet, síndrome de Ohtahara y en las epilepsias con disquinesia paroxística inducida por ejercicio. En esos casos, el test genético establecerá la etiología de la enfermedad, evitando procedimientos adicionales para la confirmación del diagnóstico y también tendrá implicancias para el consejo genético. En algunos casos, como el síndrome de Dravet y en la epilepsia con disquinesia paroxística inducida por ejercicio, también permitirán la optimización precoz de la terapia antiepiléptica.
En las EMPs, el diagnóstico molecular es indicado ante la sospecha de encefalomiopatías mitocondriales y de la enfermedad de Unverricht-Lundborg. En las encefalomiopatías mitocondriales, la confirmación diagnóstica es realizada clásicamente mediante una biopsia muscular, no obstante una biopsia negativa para las fibras rojas rasgadas no excluye totalmente el diagnóstico de MERRF o MELAS (28). De esta forma, el análisis molecular del mtDNA puede significar una confirmación diagnóstica de manera precisa y poco invasiva, así como detectar la presencia de portadores asintomáticos de enfermedades mitocondriales (25).
Consideraciones finalesLa identificación de los genes causales o que influencian el riesgo de epilepsias tiene importantes implicancias en la pesquisa y en la práctica clínica de las epilepsias. En el contexto de la pesquisa, el estudio de los efectos neurofisiológicos y del desarrollo neurológico de las mutaciones de genes identificados, pueden dilucidar los procesos básicos subyacentes a la susceptibilidad a las crisis. Esta información puede llevar al desarrollo de nuevos tratamientos dirigidos a intervenir los mecanismos específicos o a formas de prevención de la epileptogénesis. En la práctica, la utilización de la información genética puede ser utilizada, tanto para clarificar el diagnóstico en personas que ya saben que padecen, o en aquellas en las que se sospecha una epilepsia (7). Adicionalmente, una de las áreas más promisorias de la pesquisa genética de las epilepsias es la fármaco-genómica, que consiste en la búsqueda de variantes genéticas asociadas a la eficacia y a la tolerancia a los tratamientos. El uso de pruebas genéticas en variantes asociadas a respuesta a tratamientos, podrían tener beneficios clínicos evidentes (7).
Las nuevas tecnologías en el área de la genética molecular han permitido, por ejemplo, la genotipificación a gran escala de SNPs (polimorfismos de base única) y el secuenciamiento paralelo en masa. El análisis de variaciones del número de copias génicas, las llamadas CNVs (del inglés copy number variation) forman parte de este progreso en los estudios moleculares. Fanciulli y colaboradores, el 2012, identificaron micro-deleciones en el gen LGI1 a través del análisis de CNVs en familias con epilepsia del lóbulo temporal autosómico dominante con síntomas auditivos, que eran negativas para mutaciones puntuales en el secuenciación directa de los exones de ese gen (29).
La secuenciación de nueva generación ha permitido la identificación de nuevos genes en epilepsias esporádicas, caracterizadas por crisis de difícil control y combinación de retrasos en el desarrollo, encefalopatía epiléptica entre otras (30). A través de la secuenciación completa de exoma (WES – whole exome sequencing) de 10 tríos compuestos de padres no afectados y un hijo con epilepsia esporádica, en 20013 Veeramah y colaboradores encontraron mutaciones en genes conocidos o con posible significación en la excitabilidad neuronal. Cuatro de ellos tenían mutaciones en genes previamente descritos en pacientes con epilepsias graves de inicio precoz (dos en SCN1A, uno en CDKL5 y otro en EEF1A2). En tres niños las variantes estaban en genes con funciones que son posiblemente relevantes en epilepsias (KCNH5, CLCN4 y ARHGEF15). Los autores sugieren que WES sería de utilidad en el diagnóstico genético molecular de epilepsias esporádicas en niños, especialmente en crisis de inicio precoz y de difícil control (30).
La identificación de genes involucrados en la etiología de las diversas formas de epilepsias, no sólo expandirá el conocimiento acerca de las vías moleculares involucradas en la epileptogénesis, sino que además podría tener una gran repercusión en el diagnóstico, pronóstico y tratamiento de las crisis.
Los autores declaran no tener conflictos de interés, con relación a este artículo.