



INTRODUCCIÓN
Laexpiración de patentes de algunos fármacosinmunosupresores ha puesto de actualidad el tema de lasespecialidades farmacéuticas genéricas (EFG) en eltratamiento del trasplante y de las enfermedades autoinmunes. LasEFG deben satisfacer los mismos estándares de calidad,eficacia y seguridad que los aplicables al producto original,basándose en criterios técnicos establecidos por lasdistintas administraciones nacionales e internacionales(1).
La ciclosporina(CsA), uno de los principales fármacos inmunosupresores,presenta un perfil farmacocinético complejo que se traduceen una amplia variabilidad farmacocinética intra einterindividual. La monitorización de los niveles de CsApermite individualizar la pauta posológica con el objeto demantener las concentraciones sanguíneas dentro del estrechoámbito terapéutico del fármaco (2). Dado quela CsA es uno de los fármacos de dosis crítica (DC)del que se dispondrá de genéricos en un futuropróximo, la comunidad científica se plantea laidoneidad de las pruebas estándar de bioequivalencia paraEFG en medicamentos de estas características (3).
El objetivo deeste trabajo es revisar los criterios de equivalenciaterapéutica, elección de la formulación dereferencia y su aplicación a las característicasfarmacocinéticas y farmacodinámicas de laCsA.
EQUIVALENCIATERAPÉUTICA Y BIOEQUIVALENCIA
Dos productosfarmacéuticos son bioequivalentes cuando presentan unaformulación galénica equivalente (idénticacantidad de constituyentes en la misma composiciónquímica, forma y vía de dosificación, tiempode desintegración y velocidad de disolución) y susbiodisponibilidades (tasa y extensión) son similares tras laadministración en la misma dosis molar (4).
Sin embargo, labioequivalencia no necesariamente se traduce en equivalenciaterapéutica. Los fármacos terapéuticamente (esdecir, clínicamente) equivalentes se definen como aquellosque son farmacéuticamente equivalentes y tras laadministración de la misma dosis molar sus efectos respectoa eficacia y a seguridad son esencialmente los mismos(5).
Pruebas debioequivalencia
Los estudios debioequivalencia se realizan administrando a voluntarios sanos, deforma cruzada y aleatorizada, el producto nuevo y el de referenciay determinando las concentraciones sanguíneas delfármaco en función del tiempo. Se calculan lossiguientes parámetros farmacocinéticos:concentración máxima (Cmáx) ytiempo en que se produce ésta (Tmáx), comoexponentes de la tasa de liberación del fármaco yárea bajo la curva (AUC) como indicativo de labiodisponibilidad del mismo. Para algunos productos cuyaabsorción depende de la dieta deben realizarse estudios enayunas y en presencia de alimentos (1, 6).
Según lasnormativas europea y americana (FDA), dos especialidades seconsideran bioequivalentes cuando el 90% del intervalo de confianzade la relación entre el logaritmo del AUC del nuevo productoy el de referencia se encuentra dentro del intervalo de 0,80 a1,25. La normativa europea, a diferencia de la FDA, no especificalos límites para la relación entre lasCmáx, que en el caso de la FDA son los mismos quepara el AUC. Las diferencias en Tmáx se expresangeneralmente en términos absolutos y no requieren testestadísticos específicos (7, 8).
Varios autorespostulan que los criterios de bioequivalencia deberían serevaluados, y si es necesario modificados, en los fármacos deDC que reúnen algunas de las siguientescaracterísticas: estrecho margen terapéutico, altavariabilidad farmacocinética inter e intraindividual, faltade correlación entre la farmacocinética y lafarmacodinamia, cinética no lineal, cambiosfarmacocinéticos al pasar de dosis única amúltiple y formulaciones de liberación retardada. Enestos casos sería interesante diferenciar los conceptos debioequivalencia poblacional e individual; el primer caso hacereferencia a la elección de un medicamento genérico ode referencia en tratamientos de novo, mientras que el segundo concierne a la conversiónde pacientes controlados con el fármaco de referencia a unaespecialidad genérica (8, 9).
CARACTERÍSTICASFARMACOCINÉTICAS DE LA CsA
Existenevidencias, derivadas de estudios realizados con las preparacionesde CsA convencional (Sandimmun®) y deciclosporina para microemulsión (SandimmunNeoral®), de que los factoresrelacionados con la formulación alteran la cinéticadel fármaco (10). La formulación paramicroemulsión asegura la liberación rápida dela CsA dentro del tracto gastrointestinal y una presentaciónmás uniforme a la superficie absortiva del intestinodelgado. Esto produce una dispersión consistente delfármaco en el tubo digestivo sin la necesidad deemulsificadores adicionales como las sales biliares(11).
Al compararambas formulaciones en individuos sanos, la CsA paramicroemulsión presenta un perfil de absorción linealmás consistente y menos influenciado por laadministración concomitante de comida, laTmáx reducida y valores de Cmáxy AUC que oscilan entre un 70 y un 135% más altos que la CsAconvencional (12, 13).
De los estudiosiniciales realizados en voluntarios sanos se concluyó que unfactor de conversión de dosis de 0,6 producía valoressimilares del AUC al pasar de la formulación convencional ala microemulsión (11). Sin embargo, se apreció que enpacientes trasplantados renales estables este factor era inadecuadoy estudios posteriores constataron que las diferencias entre labiodisponibilidad de Sandimmun Neoral® ySandimmun® variaban entregrupos poblacionales. En trasplante renal el mantenimiento de lamisma dosis de CsA, aunque da lugar a Cmáx y AUC máselevadas (valores medios de un 37 y un 23%, respectivamente) enmicroemulsión, produce concentraciones predosis matinales(Cmín) comparables (14-17). En niños yjóvenes se han observado incrementos superiores deCmáx (125%) y AUC (85%) con CsANeoral® (18).
Losparámetros farmacocinéticos de la CsA enmicroemulsión presentan menor variabilidad intra einterindividual (3-22% frente al 19-45%) y mejor correlaciónentre Cmín y AUC (15, 19). En receptores detrasplante renal de novo la relación dosis-nivel seestabiliza antes, permitiendo un mayor controlfarmacocinético en el postrasplante inicial (16,17).
Las mayoresdiferencias entre ambas formulaciones se observan en elpostoperatorio inmediato de los pacientes trasplantadoshepáticos, que pueden alcanzar niveles sanguíneosadecuados de CsA sin la necesidad de su administraciónintravenosa. La absorción de CsA en microemulsión esindependiente de la presencia de bilis, presentando unabiodisponibilidad media del 156% mayor que la convencional (11, 20,21). En los pacientes con fibrosis quística, pancreatitis oenfermedad de Crohn la conversión a la formulaciónpara microemulsión corrige la malabsorción de la CsAobservada en la formulación convencional (11).
CARACTERÍSTICASFARMACODINÁMICAS DE LA CsA
Lindhom et al(22) demostraron con CsA convencional que la biodisponibilidad esuno de los factores farmacocinéticos determinantes tanto dela incidencia de rechazo agudo como de la supervivencia del injertotras el trasplante renal. La incidencia de pacientes libres derechazo y la supervivencia del injerto a un año aumentabasignificativamente cuando la biodisponibilidad era superior al 25%.Otros estudios corroboraron estos datos e identificaron los valoresbajos de AUC y de la concentración media de CsA (Cav),obtenida al dividir el valor de AUC por el intervalo dedosificación, como los predictores más sensibles dela aparición de rechazo agudo y pérdida del injerto aun año (23-25).
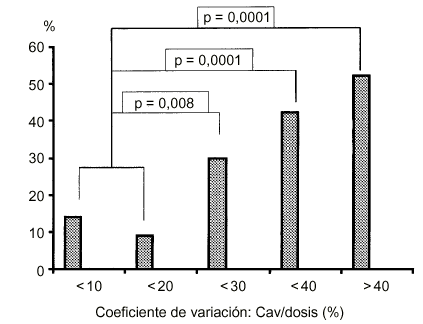
En el rechazocrónico la variabilidad intrapaciente del AUC es un factorde riesgo. Se ha demostrado que los pacientes con un coeficiente devariación de la Cav durante el intervalo de dosis superioral 20% presentan un mayor riesgo de incidencia de rechazocrónico (Fig. 1). De hecho, lavariabilidad en la biodisponibilidad de la ciclosporina parececontribuir en un 27% al riesgo total de aparición de rechazocrónico (23, 24, 26, 27).
;)
Figura 1.--Impacto de la variabilidadintrapaciente de la Cav/dosis de la ciclosporina sobre laincidencia de rechazo crónico (27). Cav:concentración media de ciclosporina.
Lacorrelación entre la variabilidad farmacocinéticaimpredecible asociada a una absorción inadecuada y un peorpronóstico clínico también se ha evidenciadoen otros tipos de trasplantes. Las fluctuaciones marcadas(superiores a un 40% de coeficiente de variación) en elnivel valle (Cmín) de ciclosporina durante elperíodo postoperatorio inicial predispone a los receptoresde un trasplante de pulmón-corazón a sufrir episodiosde rechazo durante el primer mes (28).
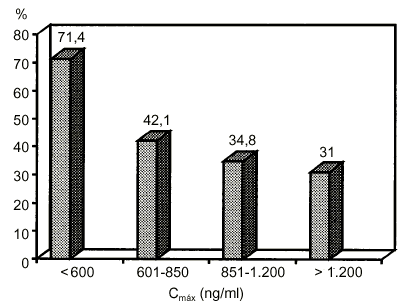
Estudiosrecientes indican que la Cmáx se correlaciona conel éxito terapéutico en trasplante renal (24),cardíaco (28) y hepático (29). En trasplantehepático el desarrollo de rechazo agudo se ha identificadocomo factor de riesgo de aparición de rechazo crónico(30). Se ha evidenciado también que los pacientes con mayorCmáx (>1.200 ng/ml) o con mayor AUC (>5.050µg/h/l) en el quinto día postrasplante presentan lamenor tasa de rechazo (28,6 y 33,3%, respectivamente), con lacaracterística adicional de no desarrollar mástoxicidad que los pacientes con valores de Cmáx oAUC menores (31). El hecho de que los pacientes que recibeninfusión intravenosa continua de CsA en el postrasplanteinmediato evolucionen significativamente peor que los tratados porvía oral revela la importancia de conseguirCmáx elevadas (32, 33).
En trasplantadoshepáticos tratados con Sandimmun Neoral® se haobservado, además, que la correlación entreCmáx y laconcentración CsA a las dos horas tras laadministración (C2) es excelente a los tres, diez yveintiún días postrasplante, que la C2 es un buenpredictor del AUC y que existe correlación entre lasconcentraciones de C2 y la incidencia de rechazo agudo, tal comopuede observarse en la figura 2(29).
;)
Figura 2.--Relación entre laCmáx de la ciclosporina para microemulsión(Sandimmun Neoral®) y laincidencia de rechazo agudo en trasplante hepático (29).Cmáx: concentraciónsanguínea máxima.
Lamonitorización de la Cmín de CsA semantiene como estándar en el manejo práctico delpaciente trasplantado a pesar de que la concentraciónpredosis no es un indicador adecuado de la exposición totaldel fármaco (34). La correlación entreCmín y AUC ha mejorado en la CsA enmicroemulsión (r = 0,80), pero la adopción demétodos con determinación de dos o másconcentraciones de CsA a determinados tiempos postdosis permitenmejorar la predicción de la exposición total. Lasestrategias de muestreo limitado que combinan las concentracionesde CsA a las cero y dos horas o a las dos y seis horas parecen serlas más adecuadas (35). En trasplante hepático ladeterminación de un solo nivel (dos horas) podría seruna alternativa válida a la monitorizaciónconvencional en ciertas situaciones clínicas (26,27).
Eficacia clínica yefectos secundarios
Lafarmacocinética mejorada de la CsA para microemulsiónha planteado la posibilidad de que sea más efectiva que laformulación convencional en la prevención del rechazoen pacientes trasplantados. Aunque no se han encontrado diferenciasrespecto a la supervivencia del injerto y del paciente, losresultados son mejores cuando rechazo agudo, pérdida delinjerto y del paciente se utilizan como una variable combinada(probabilidad estimada de 54% frente a 38%, respectivamente)(17).
Los resultadosde un metaanálisis realizado a partir de los datos demás de 7.000 pacientes sometidos a trasplante renal yhepático, estables y de novo, indican menorincidencia de rechazos al administrarse de novo y tendenciaa presentar menor número de efectos adversos(36).
En 1995, conmotivo de la sustitución de Sandimmun® porSandimmun Neoral® enEspaña, se inició un estudiofarmacoepidemiológico multicéntrico deconversión (factor 1:1) en el que participaron 1.345pacientes trasplantados renales y 296 trasplantadoshepáticos. Todos los pacientes fueron sometidos aciclosporinemias de control en los días siete y treintapostconversión, repitiéndose la determinaciónen el catorce día postconversión en caso deproducirse modificaciones de dosis. Los resultados fueronsemejantes en ambos tipos de trasplante: menor dosis de CsA con laformulación Neoral® almes de la conversión y efectos adversos comparables eincluso menores en el trasplante renal (37-39).
EQUIVALENCIATERAPÉUTICA DE CsA
Laintroducción de la CsA para microemulsión (SandimmunNeoral®) que nocumple los criterios de equivalencia respecto a la convencional(Sandimmun®) ha puesto demanifiesto la importancia de la formulación en su perfilfarmacocinético y farmacodinámico (40). La FDA en sunuevo esquema de clasificación biofarmacéuticacataloga la CsA como fármaco clase IV (baja solubilidad ypermeabilidad), que es considerada la que presenta másdificultades desde el punto de vista de la formulación (41).Dado que la absorción de la CsA es máxima en elyeyuno y prácticamente inexistente en el colon, Barr (42)postula que el tiempo del tránsito intestinal es uno de losprincipales determinantes de su biodisponibilidad. Los pacientescon tiempo reducido serían más sensibles a cambios enla liberación del fármaco de su formafarmacéutica.
Los criteriosactuales de bioequivalencia asumen que las variabilidadesintraindividuales de un fármaco son iguales y que lapoblación estudiada es homogénea, representativa ypredictiva de otros pacientes (18). En base a la experienciaadquirida, es evidente que el razonamiento es erróneo en elcaso de la CsA y otros fármacos de DC. Es posible que dosformulaciones sean bioequivalentes en la población general,pero no en subgrupos con característicasfisiopatológicas distintas (42).
Recientemente laFDA ha propuesto realizar diseños cruzados dobles en losfármacos de DC (43). La administración defármaco de referencia y genérico un mínimo dedos veces por paciente permitiría conocer la bioequivalenciapoblacional (diferencias entre los valores de bioequivalencia) y labioequivalencia individual (diferencias en la variabilidadintraindividual e interacciones sujeto-formulación). Aunqueel protocolo no está aprobado, ha recibido el apoyo de laNational Kidney Foundation (43) y se discute en el EuropeanCommittee of Proprietary Medicinal Products (44). Labioequivalencia poblacional indicaría idoneidad deprescripción, en cuanto a elección entre dosproductos al inicio de la terapia. Los criterios de bioequivalenciaindividual serían aplicables en la conversión depacientes tratados y estabilizados previamente con unfármaco de referencia (45). Johnston et al ponen de relieveque los criterios de conversión deberían asegurarseen el tiempo. Existen estudios que indican una deriva en labioequivalencia a medida que las formulaciones genéricasaumentan y que puede perderse hacia la séptimaformulación (46).
Dado que la CsApara microemulsión ha presentado mejoras evidentes respectoa la formulación convencional, debería ser utilizadacomo formulación de referencia en los estudios debioequivalencia y de equivalencia terapéutica(7).
CONCLUSIONES
La CsA es unfármaco de DC que presenta un estrecho margenterapéutico, amplia variabilidad intra e interindividual,absorción errática (formulación dependiente) yrequiere monitorización de niveles sanguíneos paraadecuar la pauta terapéutica a las necesidades del paciente.La introducción de la CsA para microemulsión hapuesto de manifiesto la importancia de la formulación en lacinética y la dinámica del fármaco.
Las normativasinternacionales deberían incluir los diseñosreplicados para estimar la variabilidad intrapaciente y detectarlas interacciones sujeto-formulación. Asimismo, lasdiferentes subpoblaciones (trasplante renal, hepático,síndrome de malabsorción, etc.) en las que se hadetectado un comportamiento diferenciado de la CsA deberíanincluirse en los estudios de equivalencia terapéutica. Encualquier caso, los niveles sanguíneos de CsAdeberían monitorizarse estrechamente tras laconversión a un fármaco genérico.
Parafármacos de DC los estudios de bioequivalenciadeberían complementarse con los de equivalenciaterapéutica. En el caso de la CsA la formulación paramicroemulsión debería ser utilizada comoreferencia.

