




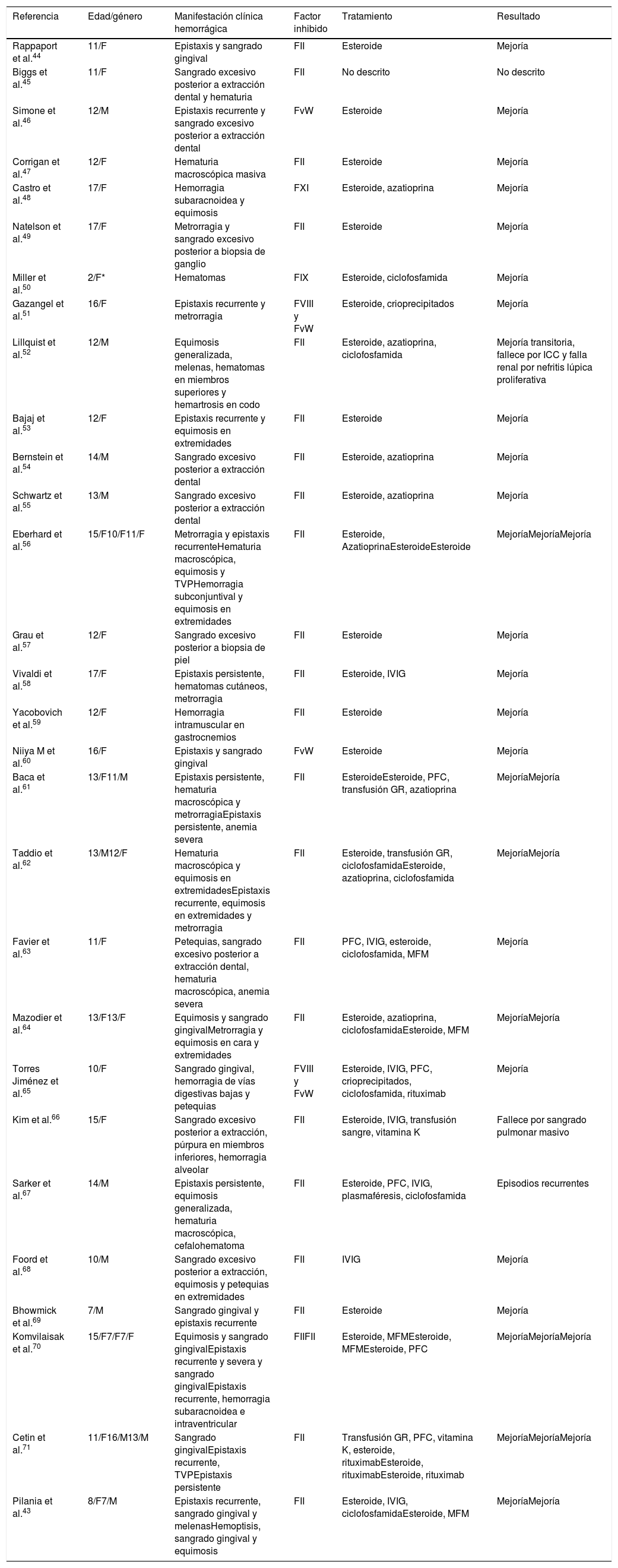
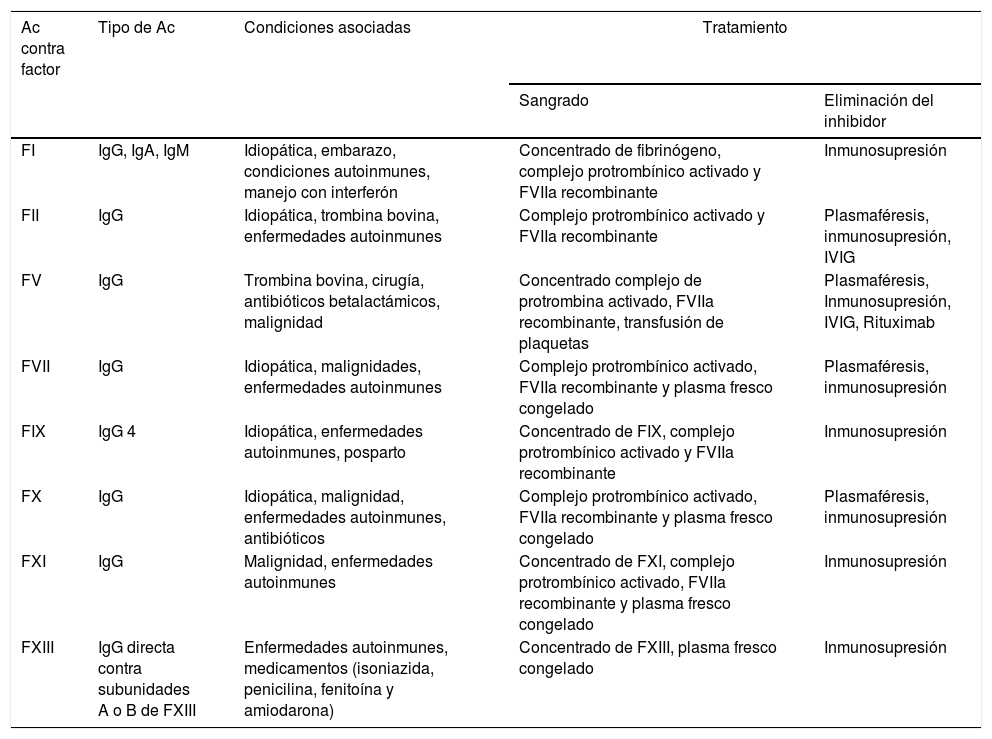
Las alteraciones hematológicas son comunes en los pacientes con lupus eritematoso sistémico (LES). Pueden expresarse relacionadas con el compromiso de las líneas celulares y con la presencia de alteraciones de la coagulación. El compromiso en la coagulación se asocia con manifestaciones trombóticas. Se han descrito factores de riesgo asociados a trombosis, como la presencia de niveles elevados de homocisteína, déficit adquirido de la proteína S, proteína C y antitrombina. Sin embargo, la diátesis hemorrágica también se ha descrito con menor frecuencia y relacionada con el déficit de factores de la coagulación, secundaria a la presencia de inhibidores. Presentamos 3 pacientes con LES juvenil con manifestaciones hematológicas poco usuales y revisión de la literatura relacionada.
Se concluye que las manifestaciones hematológicas en LES juvenil no solo se relacionan con alteraciones en las líneas celulares. Trombosis vasculares y trastornos hemorrágicos deben sospecharse. El diagnóstico precoz y el tratamiento temprano disminuyen la morbimortalidad relacionada con este tipo de manifestaciones.
Haematological alterations are common in patients with systemic lupus erythematosus (SLE). These haematological manifestations may be expressed related to the involvement of cells affected and coagulation changes. The compromise in coagulation is associated with thrombotic manifestations. Risk factors associated with thrombosis have been described, such as the presence of elevated levels of homocysteine, acquired deficit of protein S, protein C, and antithrombin. However, the haemorrhagic diathesis has also been described at a lower frequency and related to the acquired deficiency of coagulation factors caused by the development of autoantibodies directed against coagulation factors. The cases are presented of 3 patients with juvenile SLE with unusual haematological manifestations, as well as a review of the literature in relation to them.
The haematological manifestations in juvenile SLE are not only related to alterations in cell lines, vascular thrombosis and bleeding disorders should also be suspected. Early diagnosis and treatment reduces morbidity and mortality related to this type of manifestations.
Artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
