



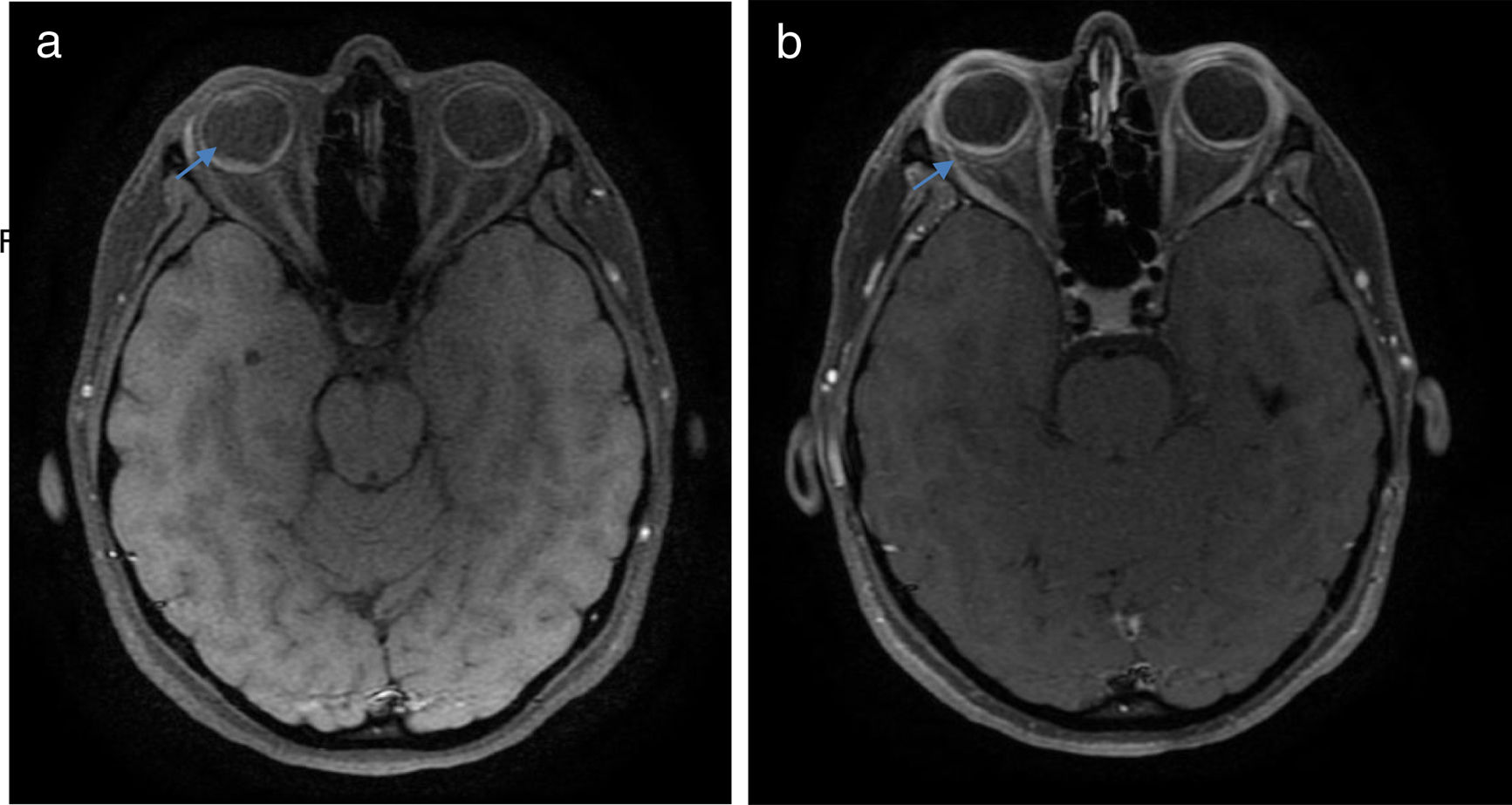
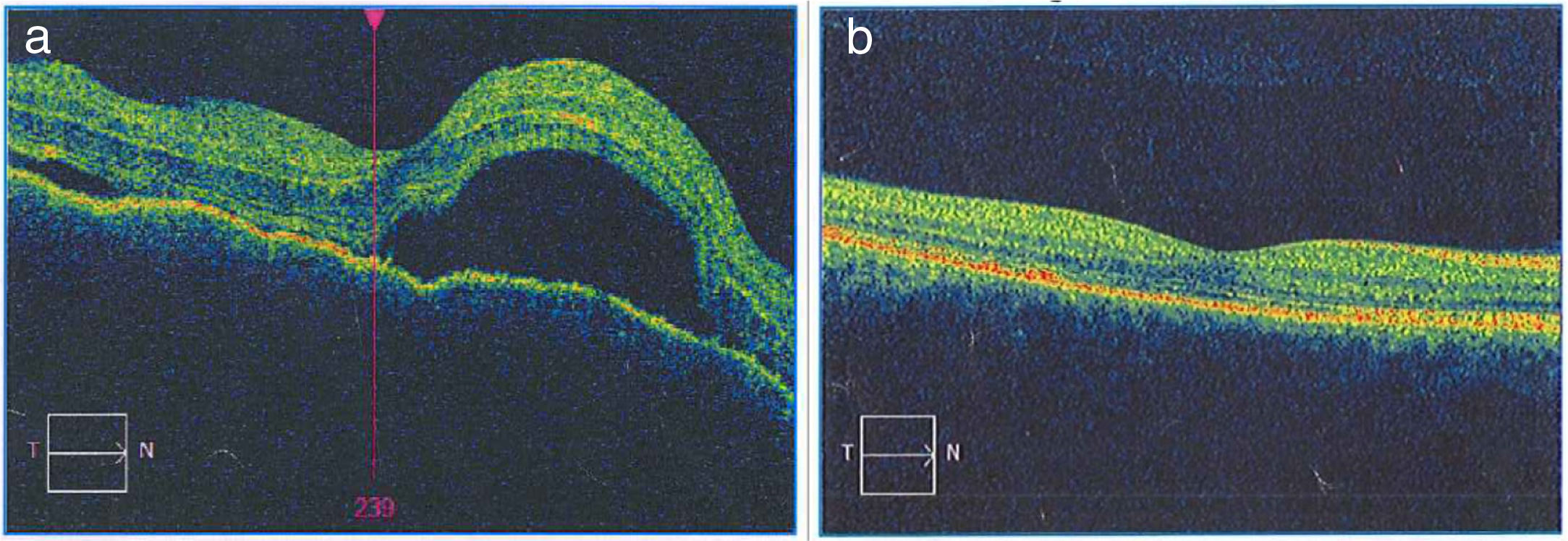
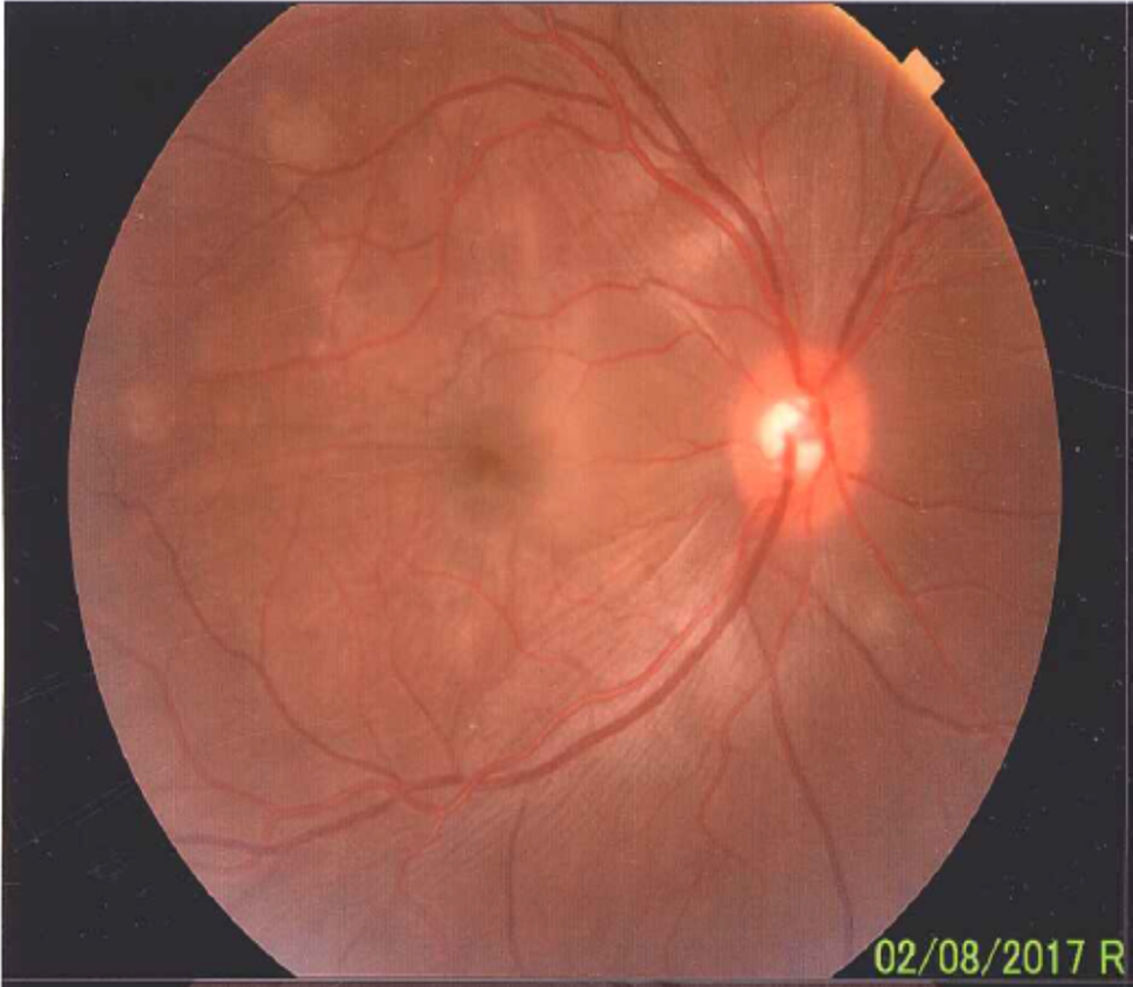
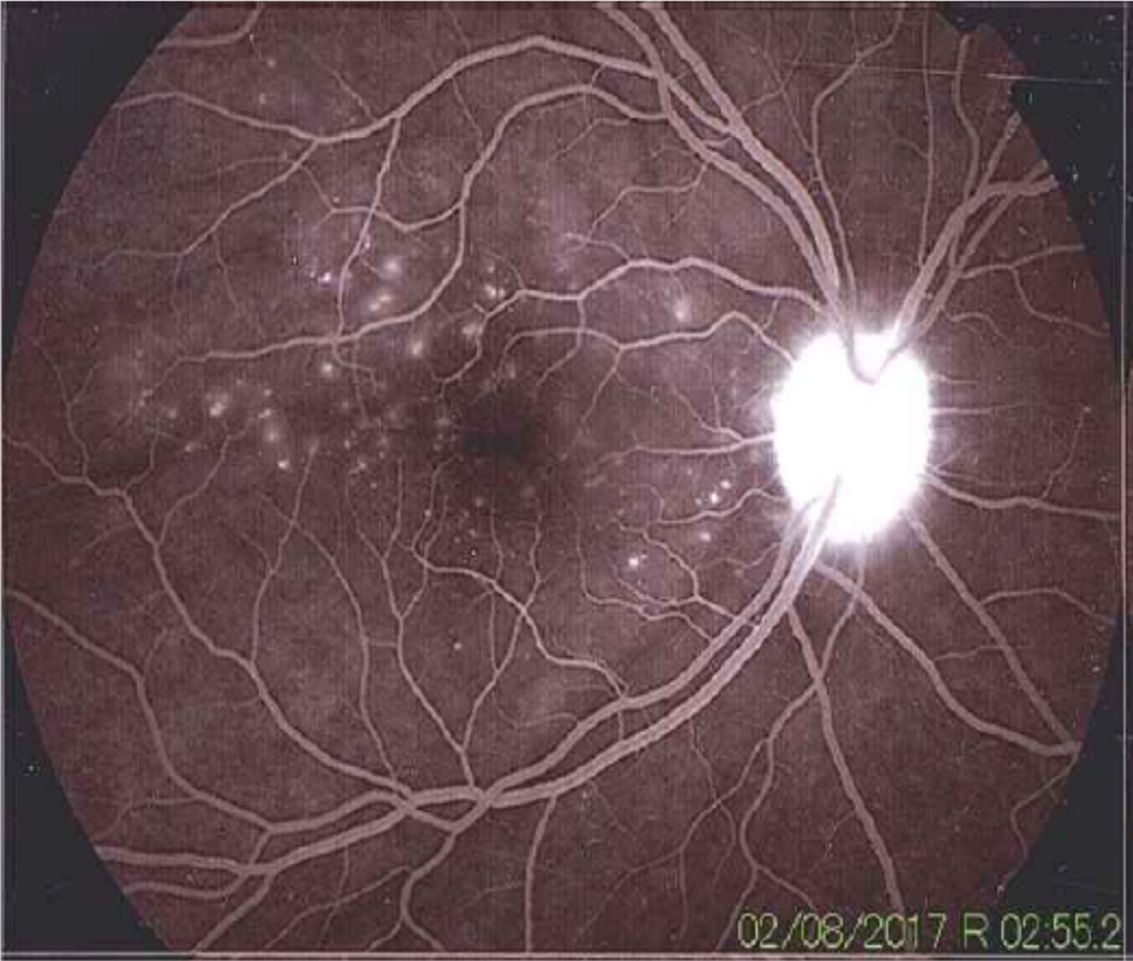
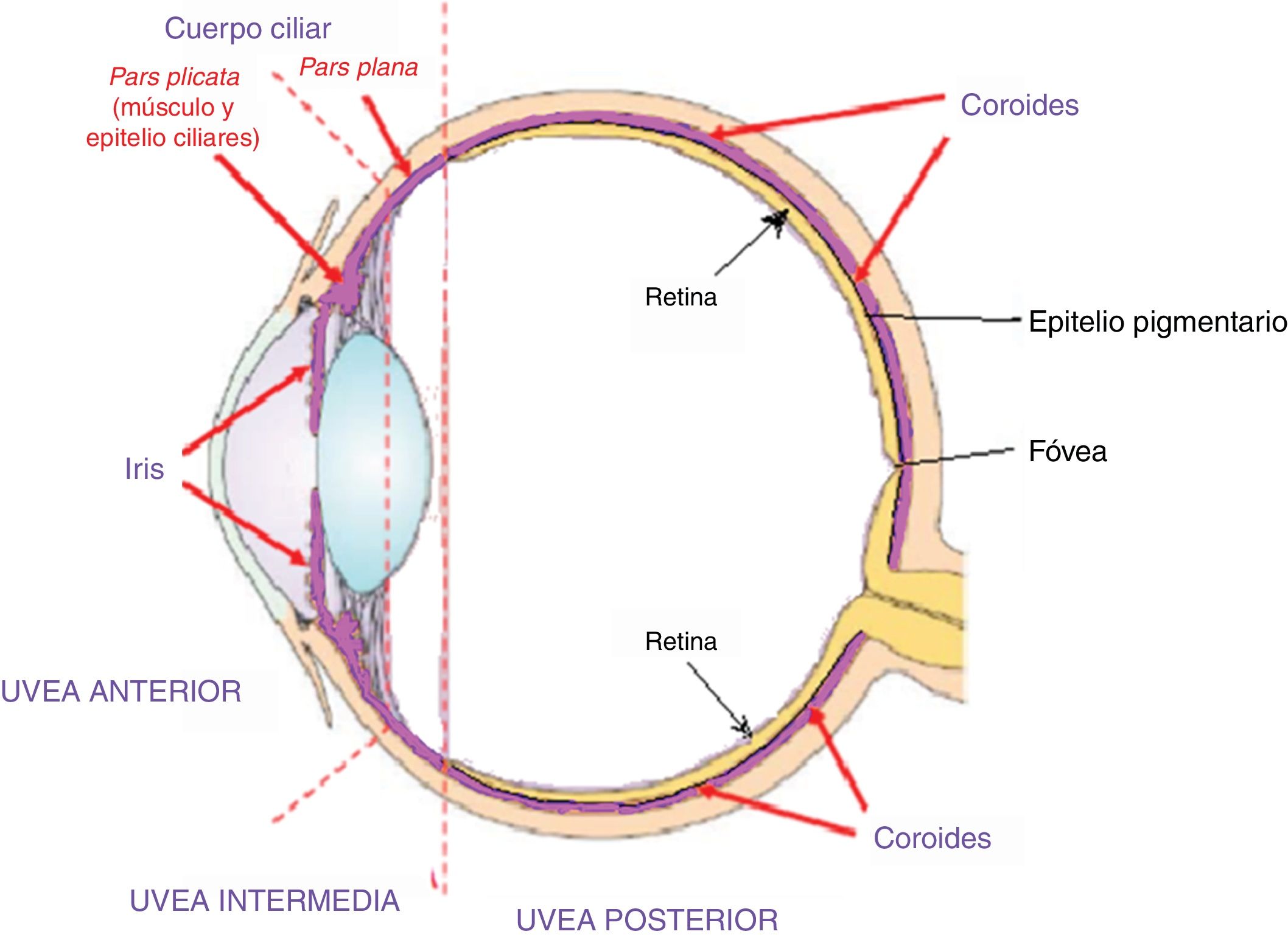
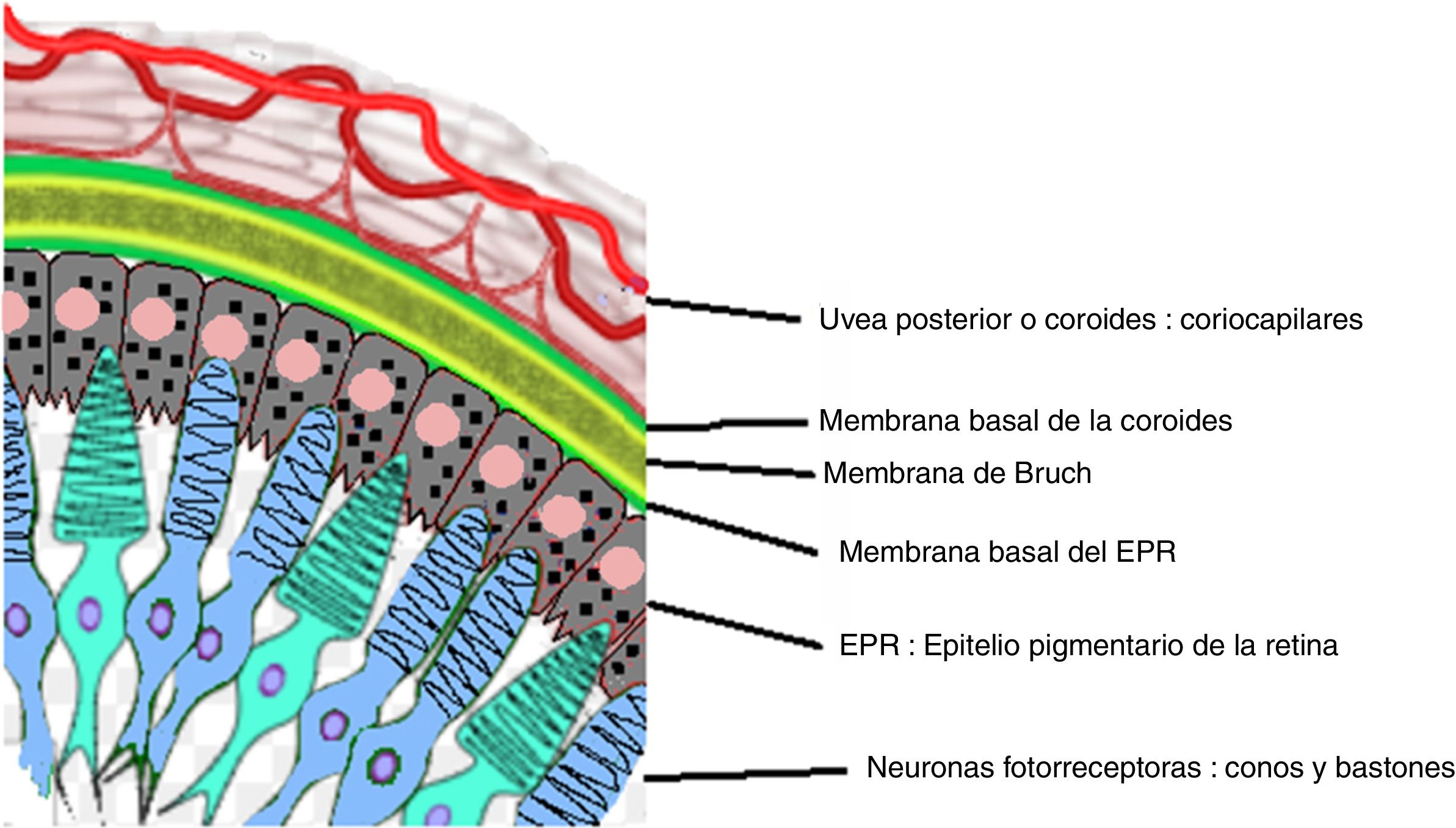

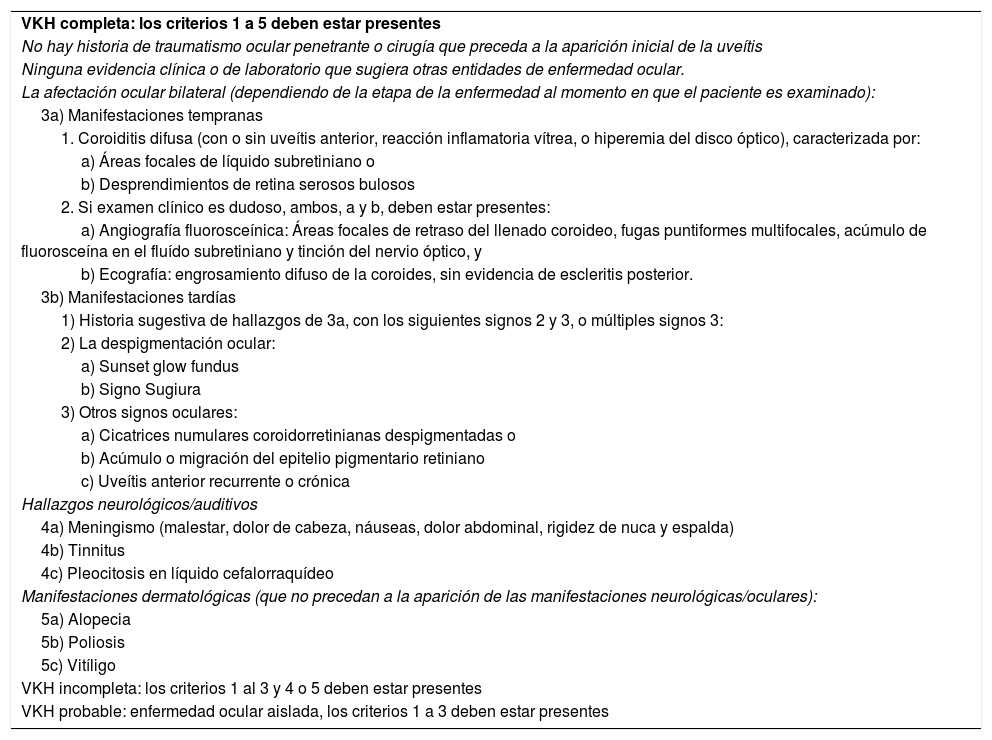
La enfermedad de Vogt Koyanagi Harada compromete múltiples órganos tales como ojos, meninges, oídos y piel. El curso progresivo de la enfermedad puede llevar a ceguera y cofosis. Se describe un caso de esta enfermedad en mujer hispana (mestiza) con alteraciones visuales, cefalalgia, tinnitus e hipoacusia a quien se le encuentra uveítis posterior con desprendimientos serosos de retina en ambos ojos y meningitis linfocitaria. El objetivo del presente estudio es, mediante una revisión de la literatura, actualizar la patogénesis inmunogenética, conocer las estrategias diagnósticas y el tratamiento apropiado.
Vogt Koyanagi Harada disease affects several parts of the body, such as eyes, meninges, ears, and skin. The progressive course of the disease can lead to blindness and deafness. The case is presented of a Hispanic woman (mixed-race) with visual alterations, headache, tinnitus, hearing loss, and posterior uveitis with serous detachments of the retina in both eyes, as well as lymphocytic meningitis. The aim of the present study is to review the literature, the diagnostic strategies, and the appropriate treatment, as well as to update the immunogenetic pathogenesis of the disease.
Artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
