





IgG4-related disease is an inflammatory systemic condition noted by the infiltration of different organs by IgG4-bearing plasma cells, as well as elevated serum IgG4 levels. Diagnosis of this condition is complex, and clinical findings are not particularly indicative. In this case series, a description is presented on 4 patients with a wide spectrum of clinical manifestations, in whom, after ruling out different options, a diagnosis of IgG4 related disease was confirmed. Despite this disease not being common, it should be considered among the options whenever multiple organs are affected. This report of patients with IgG4-related disease in Colombia highlights a wide spectrum of clinical presentations, including subglottic stenosis, autoimmune pancreatitis, retroperitoneal fibrosis, and systemic compromise.
La enfermedad relacionada con IgG4 es una condición inflamatoria sistémica, caracterizada por la infiltración de diversos órganos por complejos formados por células plasmáticas IgG4 positivas, asociadas con niveles elevados de IgG4 en el suero. El diagnóstico de esta enfermedad es complejo y los hallazgos clínicos no son patognomónicos. En esta serie de casos, describimos un amplio espectro clínico en 4 pacientes, en quienes, después de descartar otros diagnósticos, se confirmó la enfermedad relacionada con IgG4. A pesar de que esta enfermedad no es común, se debe considerar entre los diagnósticos diferenciales de enfermedades con afectación de múltiples órganos. Este reporte de pacientes con enfermedad relacionada con IgG4, en Colombia, resalta un amplio espectro de presentaciones clínicas, incluyendo estenosis subglótica, pancreatitis autoinmune, fibrosis retroperitoneal y compromiso sistémico.
Immunoglobulin G4 (IgG4) related disease is an entity described 20 years ago, initially in Japan.1 It presents itself as an inflammatory disease that can affect any organ, associated with high circulating levels of IgG4-producing plasma cells and fibrosis of the affected organs.2 Although the clinical findings may vary, the pathological findings include a lymphoplasmocytic infiltrate with abundant IgG4-positive plasma cells, phlebitis obliterans, storiform fibrosis and variable presence of eosinophils, which altogether are characteristic of the disease.3 Despite its rarity and the lack of epidemiological data, we are progressively identifying new cases in Colombia. Below we describe 4 cases of IgG4-related disease in Colombia, with a wide range of clinical manifestations, which showed a favorable evolution with the immunosuppressive treatment.
Case 1A 42-year old female patient, with no history of previous comorbidities, who consults due to a clinical picture of 3 years of evolution consisting of progressive dyspnea, from great to small efforts. In a peripheral healthcare center the patient was initially treated with a diagnosis of asthma, with conventional therapy, without any improvement. The spirometry performed at this time revealed a moderate to severe obstructive pattern without response to bronchodilator. Due to the persistence of the symptoms and the multiple visits, a computed axial tomography scan (CAT scan) of the neck and the chest was requested, which evidenced a severe tracheal stenosis (Fig. 1). The patient had no history of airway manipulation or respiratory infections. In a fibrobronchoscopy was evidenced a subglottic stenosis of more than 90% of the lumen. She was evaluated in our institution by the group of head and neck surgery, who proposed a resection of the tracheal rings and a tracheostomy. In order to rule out the diagnosis of granulomatosis with polyangiitis, a complete panel of autoimmunity tests including ANA, ENA, ANCA and rheumatoid factor were carried out, all of which were negative. In addition, the renal and hepatic function tests did not show abnormalities. The biopsy of the tracheal cartilage showed an inflammatory infiltrate which, by immunohistochemistry, showed positive staining for IgG4 in more than 10 cells per high-power field and presence of plasma cells (Fig. 2), which was consistent with a diagnosis of IgG4-related disease. Treatment with prednisone was started at a dose of 1mg/kg/day, with good clinical results. The patient was discharged without needing a tracheostomy and with resolution of the respiratory symptoms. In the clinical follow-up, 9 months after the procedure, gradual clearing of steroids was achieved, without clinical recurrence. Likewise, there was no evidence of involvement of any other organ in the follow-up.
 Coronal plane. (B) Lateral plane showing subglottic stenosis of the trachea in the cervical region. Patient 2. Magnetic resonance cholangiopancreatography. Diffuse and homogeneous alteration of the signal intensity of the pancreatic parenchyma that involves the entire head of the pancreas and the uncinate process and it extends itself to the neck of the organ.")
Imaging findings in 2 patients with IgG4-related disease.
Patient 1. Tomography of the larynx. (A) Coronal plane. (B) Lateral plane showing subglottic stenosis of the trachea in the cervical region.
Patient 2. Magnetic resonance cholangiopancreatography. Diffuse and homogeneous alteration of the signal intensity of the pancreatic parenchyma that involves the entire head of the pancreas and the uncinate process and it extends itself to the neck of the organ.
 Tracheal cartilage showing lymphoplasmacytic infiltrate and storiform fibrosis (hematoxylin and eosin, ×40). (B) Plasma cells with positive staining for IgG4, ×200). Patient 2. (A) Chronic pancreatitis with storiform fibrosis, presence of phlebitis and lymphoplasmacytic infiltrate (hematoxylin and eosin, ×100). (B) Plasma cells positive for IgG4 immunostaining (×100). Patient 3. (A) Fine-needle biopsy of the retroperitoneum, which evidences spindle cells with inflammatory infiltrate composed of lymphocytes and plasma cells (hematoxylin and eosin, ×40). (B) Plasma cells with positive immunostaining for IgG4 (×100). Patient 4. (A) Cecal appendix with circumscribed fibrosis surrounded by inflammatory infiltrate of lymphocytes and plasma cells (hematoxylin and eosin, ×40). (B) Plasma cells with positive immunostaining for IgG4 (×100).")
Pathological findings in 4 patients with IgG4-related disease.
Patient 1. (A) Tracheal cartilage showing lymphoplasmacytic infiltrate and storiform fibrosis (hematoxylin and eosin, ×40). (B) Plasma cells with positive staining for IgG4, ×200).
Patient 2. (A) Chronic pancreatitis with storiform fibrosis, presence of phlebitis and lymphoplasmacytic infiltrate (hematoxylin and eosin, ×100). (B) Plasma cells positive for IgG4 immunostaining (×100).
Patient 3. (A) Fine-needle biopsy of the retroperitoneum, which evidences spindle cells with inflammatory infiltrate composed of lymphocytes and plasma cells (hematoxylin and eosin, ×40). (B) Plasma cells with positive immunostaining for IgG4 (×100).
Patient 4. (A) Cecal appendix with circumscribed fibrosis surrounded by inflammatory infiltrate of lymphocytes and plasma cells (hematoxylin and eosin, ×40). (B) Plasma cells with positive immunostaining for IgG4 (×100).
A 54-year-old male patient, previously healthy, who was admitted to our institution referred for a clinical picture of 5 months of evolution consisting of severe abdominal pain associated to jaundice and symptoms suggestive of intestinal obstruction. Multiple imaging and laboratory studies were performed, including a nuclear magnetic resonance of the abdomen, which evidenced a lesion in the head and neck of the pancreas, highly suggestive of malignancy (Fig. 1). Within the paraclinical tests, only the transaminases were slightly elevated, whereas the levels of bilirubin, lipase, amylase and the renal function tests were normal. In view of the evidence of a pancreatic mass, the patient was taken to a partial pancreatectomy by the group of surgical oncology. The biopsy of the pancreas ruled out the presence of malignancy, while the IgG4 staining was positive in more than 10 plasma cells per high-power field, supporting the diagnosis of IgG4-related disease (Fig. 2). The immunological results for the autoimmunity panel were all negative and in the protein electrophoresis was evidenced hypergammaglobulinemia, as well as elevation of the total IgG (21g/dL, normal value up to 16g/dL) and elevation of IgG4>150mg/dL. Treatment with prednisolone at a dose of 1mg/kg/day was started, with a favorable initial clinical response, with a reduction of the abdominal symptoms and the jaundice. A progressive reduction of steroids was achieved during the clinical follow-up.
Case 3A 60-year-old male patient with a history of controlled hypertension was referred to our institution due to a clinical picture of 2 years of evolution of progressive fatigue associated with muscle pain in lower limbs, with no evidence of arthropathy or edemas of extremities. Subsequently, the patient presents exacerbation of the symptoms, with presence of intense thirst, arthralgias and myalgias of the lower limbs. The physical examination was normal. The screening for diabetes mellitus and other diseases was negative. However, the renal function tests evidenced an increase in creatinine of 2.1mg/dL (normal value up to 1mg/dL), without alterations in the urine sediment. The renal ultrasound showed the presence of a retroperitoneal mass with compressive effect on the ureters, which caused bilateral hydronephrosis that was confirmed by abdominal CT. The patient was taken to diagnostic biopsy, in which was observed the presence of retroperitoneal fibrosis with inflammatory infiltrate composed of lymphocytes and immunostaining of IgG4-positive plasma cells (Fig. 2). Additional studies ruled out the presence of malignancy. In this clinical context it was suspected the presence of IgG4-related disease and IgG subclass levels were measured for its confirmation, which showed IgG4 of 194mg/dL (normal value: 4–86mg/dL). With these findings was diagnosed IgG4-related disease (Ormond's disease). Treatment with prednisolone at a dose of 1mg/kg/day was started, with improvement of the renal function and imagenological reduction of the mass until its complete disappearance. The dose of steroids was gradually reduced up to 5mg/day, in the clinical follow-up which completed 2 years, with recovery of renal function and without relapses or commitment of other organs.
Case 4A 24-year-old male patient, who was initially evaluated in our institution by the Department of Hematology, due to a clinical picture of 5 years of evolution of microcytic anemia (hemoglobin between 7 and 8g/dL) of unknown cause, with multiple studies not compatible with hemoglobinopathies and no evidence of bleeding. The patient was hospitalized in the context of worsening of his clinical condition, with a weight loss of 10kg in 6 months, arthralgias in the knees and presence of low back pain of inflammatory characteristics. No positive findings were discovered on physical examination. The paraclinical tests on admission to the hospital showed elevation of acute phase reactants (ESR of 68mm/h, normal value up to 20mm/h), increased levels of IgG and IgM, hemoglobin of 8g/dL, low transferrin saturation and reticulocyte count. During the evaluation, the complete hematological and immunological tests, together with images, including MRI of sacroiliac joints, were normal, thereby ruling out systemic autoimmunity of the type of systemic lupus erythematosus or hemoglobinopathies. The abdominal CT scan with contrast evidenced splenomegaly and peritoneal adenopathies. In order to rule out a lymphoproliferative disorder, a laparoscopic biopsy was performed in which conglomerates of lymph nodes were found, mostly in the periappendicular region, associated with hepatosplenomegaly. In addition, an appendectomy was performed. The liver biopsy showed findings compatible with autoimmune hepatitis and the adenopathies evidenced reactive hyperplasia. The affection of the appendix showed circumscribed fibrosis surrounded by an inflammatory infiltrate of lymphocytes and the staining of plasma cells was positive for IgG4 (Fig. 2). Due to the high levels of total IgG (24g/dL, normal value up to 16g/dL), the subtypes of Ig were measured, evidencing IgG4 levels of 229mg/dL (reference value: 4–86mg/dL). With the above data, a diagnosis of IgG4-related disease was made. Due to the complexity of the disease and the systemic commitment, treatment with prednisolone was started at a dose of 1mg/kg/day with subsequent gradual decrease and rituximab (1g day 0 and 15), with adequate tolerance and marked clinical improvement, with recovery of weight, decrease in total Ig levels and improvement of the anemia. After 2 years of follow-up, the patient remains in an excellent clinical condition, with the use of 5mg of prednisolone each day and completing 2 cycles of rituximab.
DiscussionIgG4-related disease is a fibroinflammatory condition, characterized by several clinical manifestations of systemic predominance. The mostly involved organs are the salivary glands, the bile ducts, the pancreas, the retroperitoneum, the lungs, the trachea and the skin. Certain diseases that were previously categorized as different entities with their respective eponym, such as Mikulicz disease (commitment of lacrimal and salivary glands), Küttner's tumor (isolated commitment of submandibular glands), autoimmune pancreatitis and sclerosing cholangitis are now recognized as part of the spectrum of the IgG4-related disease.4,5 Due to the heterogeneity in the clinical manifestations, it is important that clinicians keep in mind this condition for the differential diagnosis of isolated or systemic commitments. With regard to the first case, with tracheal stenotic commitment, there is a report in the literature of 2 patients in whom the first manifestation of the disease was a tracheal stenosis.6 Other cases similar to our case 3 show renal manifestations defined as hydronephrosis and nephropathy associated with Ormond's disease caused by retroperitoneal fibrosis.7 In this Colombian report, special emphasis is placed on the broad spectrum of clinical and pathological manifestations, including autoimmune pancreatitis, inflammation of the cecal appendix, retroperitoneal fibrosis and tracheal stenosis.
The epidemiology of the IgG4-related disease has not been fully established. The majority of cases have been described in Japan. One study evidenced an estimated incidence of 1.08 cases per 100,000 inhabitants in 2009 and a prevalence of 0.006% for the same year, with an average age of 60 years at the time of diagnosis and a male predominance. However, the diagnosis is difficult due to the lack of familiarity with the IgG4-related disease, which is why this prevalence is poorly estimated.8
In healthy individuals, the IgG4 molecules represent at least 5% of the total IgG.9 Several immunological studies show that IgG4 does not activate the classical complement pathway and, traditionally, it has been considered to have a limited role in the immunological reaction.4 About 50% of the IgG4 molecules consist of heavy chains loosely bound by non-covalent bonds that allow their easy dissociation and formation of combinations of random chains. In this way, it allows antibodies to bind to antigens in different sites and, as a result, to lose their ability to form immune complexes.10
The diagnosis of IgG4-related disease is made by excluding different causes such as infectious diseases, systemic autoimmune diseases and lymphomas. The IgG4-related disease should be considered in case of dysfunction of one or more organs, associated with imaging, immunological and pathological findings of IgG4-related disease, for example: pseudotumors, peripancreatic inflammation, interstitial pneumonia, serum levels of IgG4>135mg/dL and biopsy with lymphoplasmocytic infiltrate, storiform fibrosis, phlebitis obliterans, eosinophilia and immunohistochemistry with positive staining for IgG4+ in ≥10 cells per high power field and a IgG4+/cells IgG4+ ratio >40%.11 The function of IgG4 in the IgG4-related disease is not sufficiently elucidated. Some authors explain it as an allergic disease, because IgG4 and IgGE are controlled by a profile of Th2 cells that produce cytokines such as IL-4 and IL-13 which increase the production of these Ig. Autoimmunity can also be considered as the initial stimulus to trigger the Th2 response in IgG4-related disease, due to the detection of autoantibodies against lactoferrin, carbonic anhydrase type ii and pancreatic trypsin inhibitor in the regular epithelium of the pancreatic, bile and salivary ducts.12
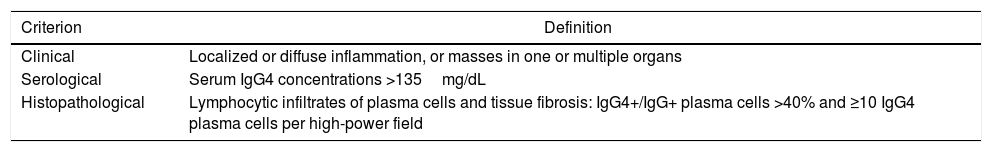
In order to unify concepts, a set of diagnostic criteria for IgG4-related disease, which includes serological, clinical and histopathological criteria was proposed in 2011 (Table 1). The definitive diagnosis is made in patients in whom all criteria are identified. A probable diagnosis is established when the clinical and histopathological criteria are met, but not the serological criteria. A possible diagnosis is established when the clinical and serological criteria are met, but without histopathological evidence.13
Diagnostic criteria for IgG4-related disease.
| Criterion | Definition |
|---|---|
| Clinical | Localized or diffuse inflammation, or masses in one or multiple organs |
| Serological | Serum IgG4 concentrations >135mg/dL |
| Histopathological | Lymphocytic infiltrates of plasma cells and tissue fibrosis: IgG4+/IgG+ plasma cells >40% and ≥10 IgG4 plasma cells per high-power field |
Patient 1 was considered with a probable diagnosis of IgG4-related disease (meets clinical and histological criteria), while patients 2, 3 and 4 were considered definitive cases (meet clinical, serological and histological criteria).
There is little evidence that supports the treatment options in IgG4-related disease. However, it is clear that when vital organs are involved, intensive treatment measures must be considered. Glucocorticoids are considered to be the cornerstone of treatment in severe cases. Without treatment, this entity progresses into fibrosis.4,14 Other immunosuppressive agents such as azathioprine, methotrexate and mycophenolate mofetil are used as maintenance therapy according to the clinical evolution. In case of refractory illness, rituximab is a useful option to reduce the levels of IgG4 and achieve clinical improvement, but further studies are needed to find better therapeutic options and improve the prognosis.15
There is still much that is unknown about this disease, especially in our country; further studies are required to establish an accurate diagnosis and appropriate therapeutic options. Immunological, epidemiological and genetic factors should be studied in our population.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflict of interestThe authors declare they do not have any conflict of interest.
Please cite this article as: Navarro E-P, Suso J-P, Chamorro M, Hormaza A, Echeverri A, Posso-Osorio I, et al. Espectro clínico de la enfermedad relacionada con IgG4 en Colombia. Rev Colomb Reumatol. 2018;25:69–74.
