










Antineutrophil cytoplasmic antibodies (ANCA) vasculitis often involves a kidney with a poor short-term prognosis. A short series of cases are presented that were treated from 2008 to 2012 in a third level hospital. Emphasis is placed on the clinical and pathological characteristics of their presentation in the emergency room, and the fact that there are no publications of a series of cases of ANCA-associated renal vasculitis.
ObjectiveTo describe the clinical outcome of ANCA-associated vasculitis in a Mexican population.
Materials and methodsA retrospective descriptive study was conducted on 23 cases of vasculitis with renal involvement.
ResultsThe study included 13 women and 10 men, with a ratio of 1.3:1, with a mean age of 47±16 years, in which 21.7% had a history of diabetes, 26% with hypertension, and 8.7% with a history of autoimmune disease. The mean duration of renal symptoms was 2.8±2.2 months, with a mean creatinine of 8.0±6.3mg/dl. The mean glomerular filtration rate was 7ml/min/m2 at admission. All (100%) of the patients had microhaematuria, and 20.9±12.2% of the patients showed dysmorphism. It is important to note that 30% of pulmonary vasculitis, 21.7% cutaneous vasculitis, and digestive tract were documented. Approximately two-thirds (65%) required renal replacement therapy on admission, a figure that remained 12 months later. There were 3 deaths associated with the activity of uncontrollable vasculitis, and only 21.7% of the patients remained free of renal replacement therapy, but with a significant deterioration in renal function at 12 months post-event.
ConclusionsANCA associated renal vasculitis has a poor short-term prognosis, and survival is closely related to the time of evolution of the disease activity and its appropriate immunosuppressive intervention.
La vasculitis de anticuerpos anticitoplasma de neutrófilo (ANCA) con frecuencia involucra el riñón, con un pobre pronóstico a corto plazo. Presentamos una serie corta de casos atendidos de 2008 a 2012, en un hospital de tercer nivel, enfatizando en las características clínicas y patológicas a su presentación en urgencias, derivado de que en México no existe una publicación sobre serie de casos de vasculitis renal ANCA asociada, únicamente casos descritos aislados.
ObjetivoDescribir el comportamiento clínico de la vasculitis ANCA-asociada en una población mexicana.
Materiales y métodosEstudio descriptivo, retrospectivo, de 23 casos de vasculitis con compromiso renal.
ResultadosLa población representada por 13 mujeres y 10 hombres, guardando una relación de 1.3:1, con una edad promedio de 47±16 años, 21.7% con antecedente de diabetes, 26% con hipertensión y 8.7% con antecedente de enfermedad autoinmune, con un tiempo promedio de evolución de sintomatología renal de 2.8±2.2 meses y una creatinina promedio de 8±6.3mg/dl que confiere una tasa de filtrado glomerular de 7ml/min/m2 a su ingreso, el 100% de los pacientes con microhematuria y un dismorfismo presente en el 20.9±12.2% de los pacientes. Es importante destacar que se documentó en el 30% datos de vasculitis pulmonar, 21.7% vasculitis cutánea y en tubo digestivo; el 65% ameritó terapia sustitutiva de la función renal a su ingreso, cifra que se mantuvo 12 meses después, con 3 defunciones asociadas a la actividad de vasculitis incontrolable y tan solo 21.7% de los pacientes permaneció libre de terapia sustitutiva pero con importante deterioro en la función renal a 12 meses posevento.
ConclusionesLa vasculitis renal ANCA asociada tiene un pobre pronóstico a corto plazo, cuya sobrevida está íntimamente relacionada al tiempo de evolución de la actividad de la enfermedad y a su intervención inmunosupresora oportuna.
Vasculitis are a group of diseases that affect the vascular wall through an inflammatory process and are traditionally classified under the Chapel Hill consensus, according to the type of blood vessel involved, as vasculitis of large vessels, medium-sized vessels and small vessels. The latter constitutes a necrotizing polyangiitis which affects predominantly venules, arterioles and capillaries; however, it may involve arteries and it can occur without any mechanism mediated by immune complexes, called for this reason pauci-immune vasculitis.1
Pauci-immune vasculitis are commonly associated with the presence of circulating autoantibodies known as anti-neutrophil cytoplasmic antibodies (ANCA) directed against antigens named proteinase 3 or myeloperoxidase which are present in the granules of neutrophils and monocytes. This type of vasculitis constitute localized clinical forms, having the glomerulus as the kidney target, developing an acute glomerulonephritis, which is called renal limited vasculitis, or may occur as a systemic form affecting multiple organs and tissues, among them the kidney, being grouped under 3 well-defined subtypes: Wegener's granulomatosis (WG), Churg-Strauss syndrome and microscopic polyangiitis (MPA). The diagnosis of these different subtypes of pauci-immune vasculitis of small vessels is facilitated based on the accompanying syndrome,2 as follows:
- -
WG occurs in association with necrotizing granulomatous inflammation of any portion of the respiratory tract.
- -
Churg-Strauss syndrome is a necrotizing granulomatous inflammation that occurs in association with asthma and eosinophilia.
- -
MPA occurs without necrotizing granulomatous inflammation, and without asthma or eosinophilia.
There are constitutional symptoms accompanying them, such as fever, myalgia, general malaise, weakness and anemia. However, they can be completely asymptomatic and their manifestations may be secondary to the deterioration in renal function in the localized form.
In Mexico, there are no publications on series of cases of ANCA-associated renal vasculitis, only isolated cases described and studies on cutaneous and respiratory vasculitis; therefore, our study derived from the Mexican population constitutes an effort to show the epidemiological and clinical behavior of renal vasculitis, with its histopathological findings and evolution.
Materials and methodsA retrospective descriptive study, in which the cases treated in the area of nephrology of a third-level hospital during 4 years, from January 2008 to December 2012 were reviewed, starting from their clinical history collected at the time of admission, their physical exploration, laboratory and paraclinical findings, including their respective immunological study, renal biopsy, therapeutic approach and clinical evolution at 12 months post-diagnosis, The inclusion criteria were:
- -
Deterioration of renal function, defined as an increase in the creatinine value >0.3mg/dl of its known basal value or a serum creatinine value >1.3mg/dl, with or without oliguria.
- -
Active urine sediment.
- -
Proteinuria in the urinalysis.
- -
Men and women of any age.
- -
ANCA positive.
- -
Renal biopsy compatible with pauci-immune glomerulonephritis.
Patients with known previous nephropathy, systemic lupus erythematosus, without compatible histological data and absence of immunological studies were excluded. Patients who lost their follow-up within the first year, irregular in their appointments and with poor adherence to the prescribed treatment were also disqualified.
The descriptive analysis was performed using measures of central tendency and ranges for the continuous variables and calculation of frequencies for the categorical variables.
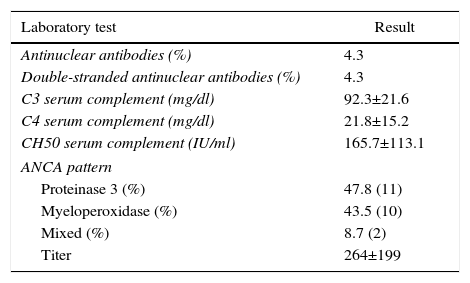
ResultsBased on the proposed inclusion criteria, 23 patients were selected between January 2008 and December 2012, 13 women and 10 men, keeping a relationship of 1.3:1, with a mean age of 47±16 years, being the youngest patient 14-year-old and the oldest 70-year old, with a mean body mass index of 26.4±5.2kg/m2, 21.7% of them with diabetes mellitus, 26.1% with systemic arterial hypertension and 8.7% with an underlying controlled autoimmune disease, type rheumatoid arthritis and primary biliary cirrhosis (Table 1). The time of evolution of their renal symptomatology at the time of consultation was on average between 1 and 3 months for 74% of the patients, 21.7% more than 3 months; in their medical history highlights the presence of constitutional symptoms in 100% of the patients, 87% with foamy urine, 78% with edema, 56.5% with oliguria, macrohematuria in 52%, weight loss referred in 35% of cases, having in around 20% data of bleeding of the digestive tract, dyspnea and hemoptysis (Fig. 1). The results of the general laboratory tests reported a mean hemoglobin of 9.8±1.9g/dl, a cholesterol level of 172.5±52.5mg/dl, triglycerides of 164.8±53.6mg/dl, serum albumin of 3.4±0.6g/dl, a serum creatinine of 8±6.3mg/dl, with a glomerular filtration rate of 7ml/min/m2, uremia of 78.7±39.9mg/dl; the analysis of urine showed microhematuria in 100% of patients, but with a mean erythrocyte dysmorphism of 20.9±12.2%, 61% with sterile leukocyturia and a mean 24-hour proteinuria of 2.3±1.5g; the renal anatomy by ultrasound reported renal lengths within normal limits. The immunological study of the patients (Table 2), included the search for antinuclear antibodies (ANA) and anti-dsDNA antibodies which were present in one of the patients, C3 serum complement with a mean of 92.3±21.6mg/dl, C4 serum complement with 21.8±15.2mg/dl, CH50 serum complement with 165.7±113.1IU/ml; ANCA antibodies were requested in all patients, finding quantitatively a titer of 264±199, whose specificities were for proteinase 3 in 47.8%, myeloperoxidase in 43.5% and with a mixed pattern in 8.7%.
General data in the population with vasculitis at admission.
| Result | |
|---|---|
| Basal characteristics | |
| Men (n) | 10 |
| Women (n) | 13 |
| Age (years) | 47±16 |
| Body mass index (kg/m2) | 26.4±5.2 |
| Systolic blood pressure (mmHg) | 141.2±29.9 |
| Diastolic blood pressure (mmHg) | 86.5±17.9 |
| Relevant antecedents | |
| Diabetes (%) | 5/23 |
| Hypertension (%) | 6/23 |
| Autoimmune disease (%) | 2/23 |
| Recurrent respiratory infection (%) | 6/23 |
| Smoking (%) | 5/23 |
| Laboratory values | |
| Total leukocytes (cell/μl) | 8900±3700 |
| Eosinophils (cell/μl) | 197±100 |
| Hemoglobin (g/dl) | 9.8±1.9 |
| Platelets (cell/μl) | 287.9±108.2 |
| Serum glucose (mg/dl) | 98.2±23.7 |
| Total cholesterol (mg/dl) | 172.5±52.5 |
| Triglycerides (mg/dl) | 164.8±53.6 |
| Uric acid (mg/dl) | 8.2±2.2 |
| Serum albumin (g/dl) | 3.4±0.6 |
| Serum creatinine (mg/dl) | 8±6.3 |
| Glomerular filtration rate (ml/min/m2)a | 7 |
| Blood urea nitrogen (mg/dl) | 78.7±39.9 |
| Erythrocyturia (%) | 100 |
| Erythrocyte dysmorphism (%) | 20.9±12.2 |
| Sterile leukocyturia (%) | 14/23 |
| Total 24-hour urine protein (g) | 2.3±1.5 |
| Urine 24-hour volume (ml) | 1370.8±1021.2 |
| Renal ultrasound | |
| Right renal length (mm) | 10.4±1.5 |
| Left renal length (mm) | 10.2±1.5 |
Values expressed as mean±SD or percentage as appropriate.

Immunological study of the population at admission.
| Laboratory test | Result |
|---|---|
| Antinuclear antibodies (%) | 4.3 |
| Double-stranded antinuclear antibodies (%) | 4.3 |
| C3 serum complement (mg/dl) | 92.3±21.6 |
| C4 serum complement (mg/dl) | 21.8±15.2 |
| CH50 serum complement (IU/ml) | 165.7±113.1 |
| ANCA pattern | |
| Proteinase 3 (%) | 47.8 (11) |
| Myeloperoxidase (%) | 43.5 (10) |
| Mixed (%) | 8.7 (2) |
| Titer | 264±199 |
Values expressed as mean±SD or percentage as applicable; values of serum complement considered as normal: C3 (70–162mg/dl), C4 (10–32mg/dl), CH50 (80–320IU/ml).
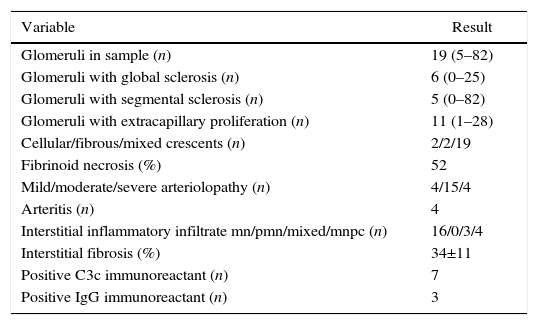
Regarding the renal biopsy study (Table 3), the average number of glomeruli per sample was 19; more than 50% of the sampled glomeruli had extracapillary proliferation, with a global sclerosis in 30% and segmental sclerosis in 26%, interstitial fibrosis in 34±11%. 100% of the biopsies had crescents: cellular 2/23, fibrous 2/23 and mixed 19/23, moderate to severe arteriolopathy in 19/23, fibrinoid necrosis in 12/23, and arteritis in 4/23 (Figs. 2 and 3).
Histopathological finding in renal biopsy.
| Variable | Result |
|---|---|
| Glomeruli in sample (n) | 19 (5–82) |
| Glomeruli with global sclerosis (n) | 6 (0–25) |
| Glomeruli with segmental sclerosis (n) | 5 (0–82) |
| Glomeruli with extracapillary proliferation (n) | 11 (1–28) |
| Cellular/fibrous/mixed crescents (n) | 2/2/19 |
| Fibrinoid necrosis (%) | 52 |
| Mild/moderate/severe arteriolopathy (n) | 4/15/4 |
| Arteritis (n) | 4 |
| Interstitial inflammatory infiltrate mn/pmn/mixed/mnpc (n) | 16/0/3/4 |
| Interstitial fibrosis (%) | 34±11 |
| Positive C3c immunoreactant (n) | 7 |
| Positive IgG immunoreactant (n) | 3 |
Values expressed as mean with its interval, absolute value or percentage as applicable; inflammatory infiltrate mn (mononuclear), pmn (polymorphonuclear), mnpc (mononuclear by plasma cells).
 Renal biopsy showing a panoramic view (10×) of a cylinder of tissue with sclerosed glomeruli (Masson")
(a) Renal biopsy showing a panoramic view (10×) of a cylinder of tissue with sclerosed glomeruli (Masson's trichrome), in which the collagen fibers that replace the normal glomerular tissue are stained with blue (b) with an extensive mononuclear and polymorphonuclear periglomerular and interstitial peritubular inflammatory reaction that ends forming a fibrocellular crescent (c) and (d).
 Glomerulus with severe interstitial and periglomerular inflammatory infiltrate (hematoxylin-eosin staining) that causes fibrin transudate forming cellular crescents that evolve to fibrocellular (b), which over few weeks end up as fibrous crescents with extensive accompanying glomerular sclerosis (c). Immunofluorescence of C3c with absence of an intense fluorescent granular pattern as in autoimmune glomerulopathies is shown, hence the term pauci-immune (d).")
(a) Glomerulus with severe interstitial and periglomerular inflammatory infiltrate (hematoxylin-eosin staining) that causes fibrin transudate forming cellular crescents that evolve to fibrocellular (b), which over few weeks end up as fibrous crescents with extensive accompanying glomerular sclerosis (c). Immunofluorescence of C3c with absence of an intense fluorescent granular pattern as in autoimmune glomerulopathies is shown, hence the term pauci-immune (d).
Of the systemic manifestations 5/23 presented pulmonary granulomatous inflammation, 2/23 with diffuse alveolar hemorrhage, 5/23 with chronic granulomatous sinusitis, cutaneous vasculitis in 5/23, as well as findings of vasculitis of the gastrointestinal tract in 5/23 (Fig. 4). Based on the clinical findings, laboratories, paraclinical exams and histology, they were catalogued as WG 10/23, microscopic polyangiitis 5/23, renal limited vasculitis 5/23 and Churg-Strauss syndrome 3/23. In the immunofluorescence was found positivity for C3c immunoreactant in 7/23 and IgG in 3/23, however, it is important to mention that not in the characteristic granular pattern by immune complexes, but rather as a stain of low intensity.

All patients received a therapeutic intervention (Table 4) requiring replacement therapy of renal function in 15/23 patients due to uremic syndrome and anuria, immunosuppressive therapy based on corticosteroids/cyclophosphamide was used in 16/23, corticosteroids/cyclophosphamide/mycophenolate mofetil in 4/23 and monotherapy with corticosteroids in 3/23, 5 patients were subjected to plasmapheresis in addition to basal immunosuppression. The final outcome of the patients with ANCA-associated renal vasculitis at 12 months was 3 deaths due to complications of the vasculitis itself, 15/23 under chronic dialytic therapy and 5/23 with dialysis-free immunosuppressive therapy, with a stage 4 chronic kidney disease (GFR 15–29ml/min/m2).
Treatment used in patients with vasculitis.
| Therapy | Result |
|---|---|
| Corticosteroids+cyclophosphamide | 16 |
| Corticosteroids+cyclophosphamide+mycophenolate | 4 |
| Monotherapy with corticosteroids | 3 |
| Plasmapheresis | 5 |
| Rituximab | 1 |
| Blockers of the renin-angiotensin-aldosterone system | 12 |
| Furosemide | 10 |
| Renal function replacement therapy: hemodialysis/peritoneal | 10/5 |
Values expressed as number of cases.
The cause of systemic ANCA-associated vasculitis is probably multifactorial, since it remains unknown to date; however, many associated factors of ethnic, geographical, environmental, genetic and of drug exposure type have been studied over the years, despite this, without a timely medical intervention it still has a high mortality rate at one year, nearly 80%, derived from the multiple complications that it causes. It has been possible to reduce such mortality with the use of the immunosuppressive therapy that allows to control the disease activity and limit the damage to the involved organs.2,3
Epidemiology of systemic vasculitisPauci-immune vasculitis has an annual incidence of about 5–20 cases per million inhabitants and varies from one region to another.4 Systemic vasculitis occur most frequently at an adult age, from the fifth to the seventh decade of life, however, pauci-immune glomerulonephritis with rapidly progressive pattern are the most common forms of crescentic GN at all ages, including elderly people. In our case series, although the average age exceeded 40 years, we had patients with extreme ages ranging from 14 to 70 years, who did not have an antecedent as it normally happens, which demonstrates that vasculitis does not discriminate on the basis of age. There is no pattern of hereditary transfer that allows the development of WG in more than one member of the family, however, associations with some HLA DR2, HLA B7 and HLA DR1 antigens have been described without any further explanation. The literature describes a higher prevalence in Caucasian population than in Afro-Americans, as well as a tendency to more cases of WG in cold climates and to MPA in warm climates.5–7 In the same way, with an uncertain unknown mechanism, the chronic contact with silica is described as an environmental risk factor for the development of small-vessel vasculitis; also, the chronic exposure to drugs such as propylthiouracil, penicilamine and minocycline has been associated with vasculitis with high titers of antibodies against myeloperoxidase (p-ANCA).8–10 In this regard, our population was of Caucasian origin and coming from all over the country, although the majority from the Valley of Mexico with an altitude which exceeds 2220meters above sea level, where the temperatures are colder; also, the close relatives of our patients were studied as part of the investigation of each case, without finding abnormalities that would justify further intervention in them.
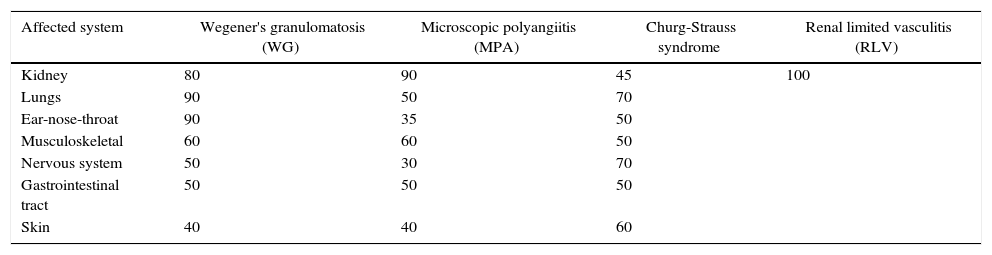
Sites affected by systemic vasculitisAlthough the first manifestations of systemic vasculitis are usually a flu-like syndrome, with myalgias, arthralgias, anorexia, weight loss, fever, malaise, etc., they do not constitute the main cause of consultation, until a specific organ results affected.11 Thus, in our patients the time of consultation was late in the majority of cases, with a time of evolution of the renal symptomatology ranging from one to three months, which marks the prognosis by the level of glomerular damage found in the biopsies, considering their pathogenic characteristics of being necrotizing and rapidly progressive due to extensive tissue destruction. Today is already known that the pauci-immune small-vessel vasculitis can affect any organ, but their frequency or proportion is variable according to the type of vasculitis (Table 5).2,12 Likewise, there is a variety of presentations reported in each affected organ or tissue (Table 6),2 we could document at least 3 sites of extrarenal vasculitis in the patients, some of them coexisting, for example: renal vasculitis with pulmonary, sinusal and gastrointestinal granulomas, which had no correlation with the titer of the anticytoplasmic antibodies or with their respective present specificities.
Sites of organ affection in systemic vasculitis (%).
| Affected system | Wegener's granulomatosis (WG) | Microscopic polyangiitis (MPA) | Churg-Strauss syndrome | Renal limited vasculitis (RLV) |
|---|---|---|---|---|
| Kidney | 80 | 90 | 45 | 100 |
| Lungs | 90 | 50 | 70 | |
| Ear-nose-throat | 90 | 35 | 50 | |
| Musculoskeletal | 60 | 60 | 50 | |
| Nervous system | 50 | 30 | 70 | |
| Gastrointestinal tract | 50 | 50 | 50 | |
| Skin | 40 | 40 | 60 |
Source: Jenette et al.2,11
Forms of presentation of systemic vasculitis in different organs.
| Organ or tissue | Manifestations |
|---|---|
| Skin | Purpuric recurrent outbreaks in lower limbs, cutaneous nodules, ulcerations. |
| Respiratory tract | Pulmonary hemorrhage, parenchymatous granulomatous lesions in the form of nodules or cavitations, sinusitis, osseous perforation of the nasal septum and “saddle” nose deformity, subglottic stenosis, rhinitis, otitis media, inflammation of the eyeball |
| Heart | Transient heart block, ventricular dysfunction, pericarditis, myocarditis, endocarditis, infarction |
| Nervous system | Multiple mononeuritis, cerebral and meningeal vasculitis |
| Gastrointestinal | Abdominal pain due to mesenteric ischemia, digestive tract bleeding, intestinal perforation, pancreatic and hepatic vasculitis manifested as pancreatitis and hepatitis, respectively |
Source: Jenette et al.2
Renal involvement in ANCA vasculitis is frequent, in general, the literature describes it in 50% of patients at the time of diagnosis and in 70% to 85% during the course of the disease.13 Renal compromise is very common in WG and MPA, and less frequent in Churg-Strauss syndrome, being hematuria, proteinuria and kidney failure the leading complications. The latter with a rapidly progressive pattern and severe characteristics in WG and MPA, about 40% to 95% of WG have renal compromise at their presentation and 64% to 100% of PAM3; in our series, 100% of the population had erythrocyturia with a dysmorphism of 20%, which is considered low taking into account the type of glomerulonephritis in question, however, the level of renal damage was high with an average glomerular filtration rate of 7ml/min/m2 and need for urgent dialytic therapy in 65% of patients, which was confirmed by the findings in their renal biopsies with the high degree of fibrosis and extensive necrosis of the glomerular vascular walls. Over time, although all patients received immunosuppressive therapy, renal survival was low, since only 21.7% (5 patients) remained free of dialysis at 12 months, maintaining a glomerular filtration rate between 15 and 29ml/min/m2, being classified within stage 4 of chronic kidney disease according to the classical classification proposed by The National Kidney Foundation Kidney Disease Outcomes Quality Initiative, which is finally a bleak prognosis in their clinical evolution in the short term.
Treatment optionsThe treatment should be guided by the premise of “not overtreat a mild illness, or undertreat a severe illness”; the indication for a complete immunosuppressive treatment is a severe glomerulonephritis that causes deterioration in the renal function, so treatment involves 3 phases: induction to remission, maintenance of remission and treatment of relapse.14
With the introduction of the alkylating agents such as cyclophosphamide, the prognosis improved with the increase of renal survival from 20% to 60% at one year with corticosteroids alone, to be up to 87% at 8 years and 64% at 10 years, therefore, the most effective regimes for induction to remission currently include both drugs.3,15,16 Once remission is achieved, drugs with less toxicity such as azathioprine, methotrexate, mycophenolate mofetil and cyclosporine are used to maintain remission. The relapse rates are lower in MPA than in WG.
In our study population, all patients were submitted to the first phase of induction of immunosuppression, but not all were candidates to continue it, especially after performing the renal biopsy and reclassifying each patient according to the histological findings, subtracting prognosis of their renal function, which forced us to reduce the immunosuppression and later discontinue it. In this way, the patients received a different scheme of immunosuppression defined based on the clinical and histological criteria, some of them received only corticosteroids (3/23), others received corticosteroids combined with cyclophosphamide-type alkylating agents (16/23) and mycophenolate mofetil was added in addition to the foregoing as a third drug to others (4/23), here is important to mention that we had access to therapies such as plasmapheresis and rituximab, which were indicated according to the assessment by consensus of the specialists in nephrology of the institution.
Factors of poor prognosis in ANCA renal vasculitisThe accepted prognostic factors are the rate with which treatment is started, the renal function at the time of diagnosis and the extension of the commitment in the renal biopsy.13 In the last decade, other additional factors that confer to ANCA vasculitis a bleak prognosis have been studied and defined, among them, the presence of ANCA antimyeloperoxidase (p-ANCA) associated with greater interstitial fibrosis, tubular atrophy and glomerular sclerosis,17 in this way, the greater glomerulosclerosis and interstitial fibrosis in renal biopsy, the worse renal prognosis in the long term, despite receiving intensive immunosuppressive treatment including plasmapheresis.15
In the patients treated by us, we considered that their evolution at 12 months was the result of a coexistence of several of these factors: tubulointerstitial inflammation, mostly mononuclear (87%), high fibrinoid necrosis and moderate to severe arteriolopathy (83%), high proportion of glomeruli with extracapillary proliferation, as well as segmental and global sclerosis (57%) and interstitial fibrosis of at least 35% of renal tissue. This without taking into account clinical criteria such as the age of the patient, underlying comorbidities and the time of evolution of the glomerulonephritis without receiving immunosuppressive intervention; we consider that the last one played an important role in the perpetuation of the inflammatory state of the renal tissue, causing glomerular sclerosis and extensive interstitial fibrosis, leaving little or no viable tissue at the moment of the intervention, which obviously reduces the renal survival despite using novel measures such as monoclonal antibodies and plasmapheresis.
There is a study in which histological and clinical criteria are established as predictors of poor evolution toward a definitive dialytic therapy at the time of admission and at 12 months, thus, in multivariate analysis there is a negative correlation to present greater deterioration of renal function with the age of the patient, tubular atrophy, fibrous crescents, acute and chronic tubulointerstitial damage; in the same way, it was found an association of better renal function and dialytic independence at 12 months with a higher percentage of normal glomeruli found in the renal biopsy and the use of plasmapheresis as a treatment option.18
Kidney transplantation in ANCA vasculitisWith the onset of renal failure and dialytic therapy many glomerulonephritis reduce their activity to the point of disappearing; however, some continue being active and go unnoticed increasing the associated mortality; for this reason, patients should be monitored.19 Once transplanted, patients who are maintained under a regime based on prednisone, mycophenolate and cyclosporine or tacrolimus, have a satisfactory evolution, with good prognosis and high survival rates for them and their grafts. The recurrence of active glomerulonephritis in the renal graft is between 15% and 37% and respond well to therapy with cyclophosphamide and other more intensive therapies.2,13
In our series, 3 patients underwent successful kidney transplantation, without subsequent complications of relapse in more than 4 years of evolution with their grafts, it is important to mention that other patients are currently on transplant protocol, but it has not been possible to accomplish the surgery for reasons other than the etiology of the chronic kidney disease or consensus of the transplant team, having in mind that the antecedent of the vasculitis is not a contraindication for the transplant.
ConclusionsANCA-associated renal vasculitis has a poor prognosis in the short term, survival is related to the time of evolution of the disease activity and the timely immunosuppressive intervention, so that early detection through intentional search on the slightest suspicion is transcendental in the improvement of the prognosis of this type of patients.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingThe authors declare they have done the work with funds of the lead author.
Conflict of interestThe authors declare they do not have any conflict of interest.
Please cite this article as: Polanco NA, Soto-Abraham M, Vázquez Rangel A. Características clinicopatológicas de la vasculitis renal pauciinmune en México: reporte de 23 casos. Rev Colomb Reumatol. 2017;24:70–78.
