


Se describe la relación funcional del metabolismo de las grasas y los hidratos de carbono y su interdependencia, desde los tradicionales conceptos del ciclo glucosa-ácidos grasos (Randle) y la hipótesis portal de la insulinorresistencia hasta los nuevos sobre los adipocitos marrones y beiges, con énfasis en el normal funcionamiento de un patrón endocrino cuya disfunción es clave en la fisiopatología: el eje adipoinsular, vinculado funcionalmente incluso con el hipotálamo, la hipófisis y las adrenales, que involucra 2hormonas adipogénicas (insulina y glucocorticoide) que facilitarían el desarrollo de la grasa omental perivisceral, con fuertes consecuencias metabólicas. Se discute la ectopia o asiento de grasa en tejido magro por incapacidad del tejido adiposo para seguir atesorando grasas y la actividad endocrina del adipocito, con la producción de moléculas que influyen sobre los mecanismos productores de insulinorresistencia (leptina, adiponectina, TNF-α, resistina, etc.) y disfunción insular. Se describe la disminución de la capacidad oxidativa en la cadena respiratoria mitocondrial y el renacer del concepto de lipogénesis de novo, ambas favorecedoras del atesoramiento de grasas intracelular. En tejidos magros existen pequeñas reservas intracelulares de grasas que mantienen una regulación de funciones esenciales, aunque si aparece una sobrecarga lipídica, el fenómeno conduciría a disfunción (lipotoxicidad) y muerte celular (lipoapoptosis). La tormentosa relación entre las grasas y el islote de Langerhans va más allá del esfuerzo funcional que impone la insulinorresistencia periférica sobre la célula β, por efectos directos de los lípidos o sus derivados sobre la función del islote pancreático. Sin déficit de insulina no hay diabetes.
A review is presented on a functional relationship between fat and carbohydrate metabolism and inter-dependence from the traditional concepts of glucose-fatty acids cycle (Randle), and from the insulin resistance portal hypothesis up to the new aspects on brown and beige adipocytes. Emphasis is placed on the normal function of an endocrine pattern, in which its malfunction is the key in the pathophysiology of these conditions: the adipoinsular axis, with a functional link with the hypothalamic-pituitary-adrenal axis, which involves 2adipogenic hormones (insulin and glucocorticoid). This has an influence on the development of omental peri-visceral fat, with severe metabolic consequences. A discussion is also presented on the concept of ectopic fat on non-adipose tissues that results in the incapacity of fatty tissue for storing lipids and the considerations about the endocrine activity of adipocyte producing substances that influence several mechanisms that could result in insulin resistance (leptin, adiponectin, TNF-α, resistin, etc.). New aspects are considered regarding the decrease in the oxidative capacity in the mitochondrial respiratory chain, and the re-birth of the concept of de novo lipogenesis that increases the storing of intra-cellular fat. In non-adipose tissues there are small intra-cellular fat quantities for essential functions, but lipid overloading leads to cell dysfunction (lipo-toxicity) and death (lipo-apoptosis). The stormy relationship between fat and Langerhans’ Islets goes beyond the functional effort as consequence of peripheral insulin-resistance and the pancreatic beta cell suffers a direct lipid (or derivatives) functional effect. Without insulin deficiency diabetes does not appear.
La epidemia de obesidad y de diabetes de tipo 2 (DMT2), que en principio se llamó «melliza», tiene en realidad un sutil tercer integrante: la resistencia a la insulina. Por eso es, en realidad, una «epidemia trilliza». Ello no conforma un concepto menor, ya que el crecimiento asombroso de estos problemas condiciona la salud pública mundial, pues las afecciones metabólicas matan a través de la enfermedad cardiovascular. Por eso son «asesinos silenciosos».
Las investigaciones han sido esenciales para desarrollar la idea de asociación entre la obesidad y la DM. Así, progresivamente, la DM no solo se vinculó a la severidad de la obesidad, sino a la ganancia de peso, la duración de la ganancia de peso, el tipo clínico y también a la evidencia de la prevención o la mejora del trastorno metabólico cuando la persona obesa puede adelgazar.
La resistencia a la insulina como componente esencial en la etiopatogenia de la DMT2 se ha definido tradicionalmente desde el punto de vista glucotóxico. Con el pasar de los años se gestó y se fortaleció la idea de que las grasas influyen de manera decisiva sobre el metabolismo de los hidratos de carbono, sobre la actividad de la insulina y, a través de ellos, en el desarrollo de la DMT2. Existen numerosos trabajos experimentales y de investigación epidemiológica y clínica que cimentaron una interesante historia que, en parte, muestra la tormentosa relación entre la obesidad y la diabetes. Los ácidos grasos libres (AGL) y la glucemia. El tejido adiposo y los islotes de Langerhans.
Reaven opinó que, a pesar de la intensa búsqueda, aún no es clara la base fisiopatológica de la DMT2 y que ello ha dado origen a numerosas hipótesis y especulaciones, e incluso a revisiones como la de McGarry, quien luego de un fascinante título, interpretó desde un ángulo alternativo el origen de la DMT2, lo que provocó un sinnúmero de controversias. En ese artículo que publicó Science en 1992 se preguntó: ¿Qué habría sucedido si Minkowski hubiese sido agéusico? Allí, exploró los conceptos que representan la visión «tradicional» centralizados en la hiperglucemia y propuso que los fenómenos de resistencia a la insulina y de hiperglucemia se podrían entender mejor si se vieran en el contexto de las anormalidades lipídicas subyacentes1.
Del mismo modo, en los últimos años ha cambiado de forma drástica la opinión que atribuía a los adipocitos la característica de tejido relativamente inerte para guardar energía. Se han hallado y se les atribuyen tantas actividades que es difícil reunir todas sus funciones, que los muestra, además, como una verdadera glándula endocrina2 con un desempeño vital en el metabolismo. El adipocito como órgano dinámico regula finamente el balance nutricional a través y por medio de un complejo intercambio con los órganos y el medio que lo rodea (es decir, desarrollando actividad endocrina y, además, actividades paracrina y autocrina).
Existen defectos génicos y efectos ambientales difíciles de determinar, pero capaces de inducir desde la acumulación de grasa central abdominal y en tejidos no adiposos (ectopia grasa) hasta la movilización de lípidos y la producción de diversas sustancias que se originan en el tejido adiposo, pero que influyen en la función de otros órganos, todo como reflejo de un fenómeno de mala adaptación del metabolismo energético. Tal vez, los cambios en los lípidos séricos y tisulares se constituyen en los mayores perpetuadores de la insulinorresistencia. No se conoce cuál es el fenómeno original, pero tradicionalmente se considera que las alteraciones en el intercambio de glucosa y de ácidos grasos (AG) en el músculo y la pérdida la capacidad para suprimir la liberación de AG y glicerol desde los adipocitos son fenómenos tempranos en la disfunción metabólica3–5. Las líneas de investigación se amplían permanentemente. Desde la lipogénesis hasta la oxidación de grasas y el estrés de organelas celulares, el efecto tóxico cuando circulan en exceso o se ubican en lugares que no se encuentran preparados para guardar grasas son motivo de profundas inquietudes y de estudios.
Todo fundamenta la revisión de aspectos cardinales del vínculo entre las grasas y la DMT2: la influencia de los AGL, los conceptos sobre grasa central y ectópica, el paradigma del adipocito secretor y los hallazgos de novedosas evidencias sobre mecanismos que intervienen en la patogenia de la DM. Asimismo, es imperioso reconocer que el tejido adiposo es esencial en la regulación del gasto energético —por tanto, para la vida— con la propuesta de que dentro de la homeostasis existiría un eje adipoinsular. El problema es cuando se rompe el equilibrio y el adipocito de amigo, pasa a enemigo.
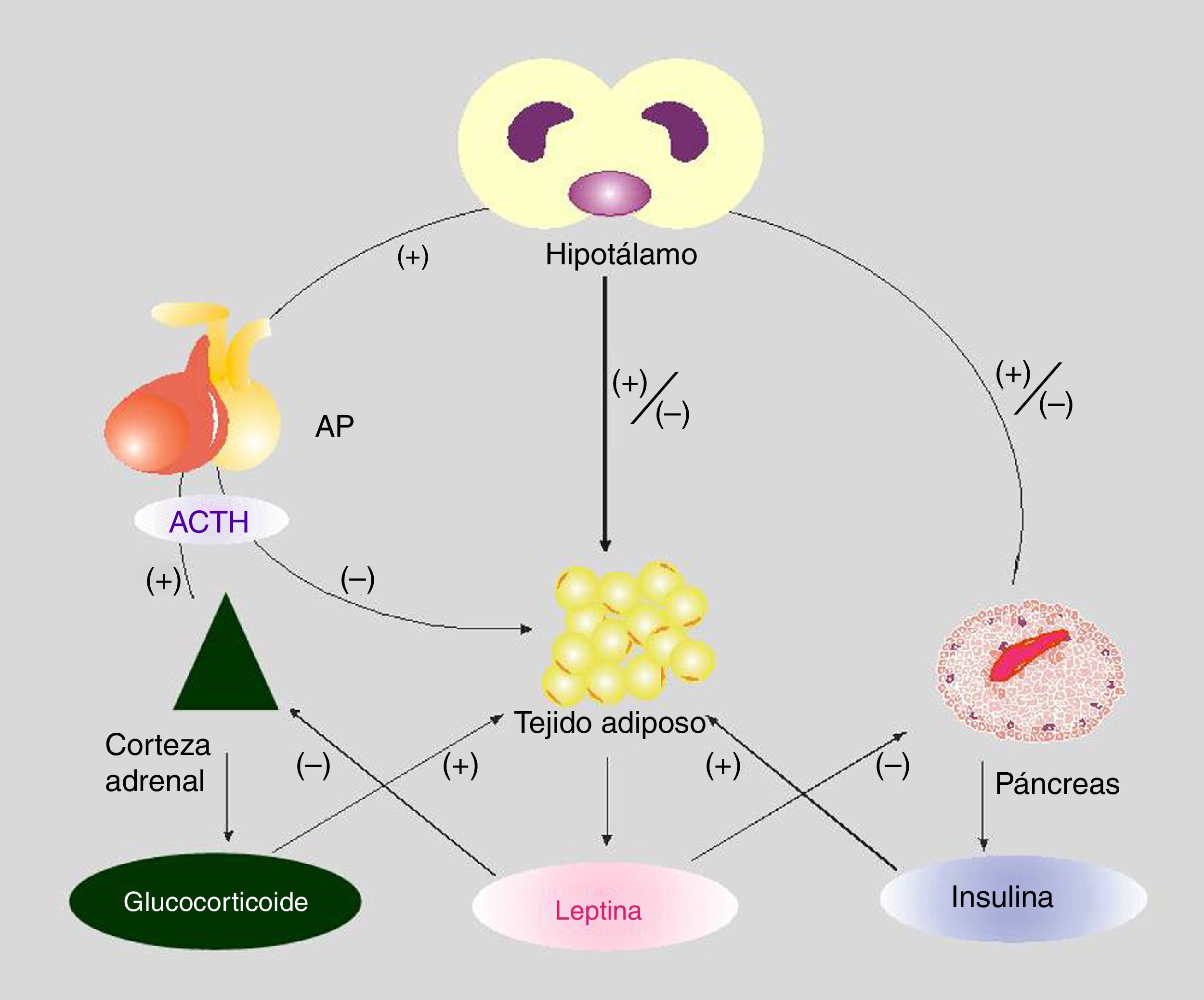
Interacción entre los ejes hipotálamo-hipofisocorticoadrenal y adipoinsularSe ha propuesto la existencia de un eje adipoinsular6, que se organiza a partir del efecto estimulador de la insulina —secretada por la célula β— sobre la liberación adipocitaria de leptina, por un lado, y por la actividad inhibidora de la leptina circulante sobre la síntesis de insulina, por otro, ya sea directamente sobre la célula β pancreática o, indirectamente, mediado por el sistema nervioso autónomo (SNA).
La leptina es una hormona proteica de cadena simple de 167 aminoácidos y constituye la adipocina que más se ha estudiado y que se produce y secreta a partir del gen ob, cuyos niveles circulantes correlacionan directamente con el índice de masa corporal (IMC), debido a que, mayormente la masa grasa, determina la masa corporal. Sin embargo, los niveles de leptina aumentan luego de una ingesta y disminuyen en el ayuno, sin que ocurran cambios apreciables en la masa del tejido adiposo blanco (TAB)7.
El receptor de leptina Ob-R, que se distribuye en la mayoría de los tejidos, forma parte de la familia de receptores de clase I de citocinas y se expresa en distintas isoformas (Ob-Ra-f). La isoforma «larga», Ob-Rb, posee todos los dominios implicados en la señalización intracelular y activa factores de transcripción del tipo signal transducer activator transcription (STAT). Este aumenta la expresión en distintos genes, entre ellos el supresor of cytokine signaling que, en turno, bloquea la vía de señalización estimulada por Ob-Rb por un mecanismo de «feed-back» negativo intracelular8.
La insulina actúa directamente sobre el adipocito por aumento de la expresión de Ob y la secreción de leptina in vitro, efectos que requieren cierto tiempo para manifestarse in vivo, no siempre ocurren en forma aguda y en condiciones fisiológicas y además, dependerían de los glucocorticoides9. A su vez, la leptina modifica la función de la célula β pancreática y su administración in vivo disminuye la insulinemia en animales alimentados y ayunados. Los estudios in vitro, aunque controversiales, demuestran un efecto inhibitorio directo de la leptina sobre la liberación de insulina basal y posglucosa10. La inervación pancreática por el SNA contribuye al efecto inhibitorio, la administración de la leptina in vivo inhibe la expresión y secreción del neuropéptido Y hipotalámico, que disminuye el tono del sistema nervioso parasimpático y el estímulo autonómico sobre la secreción de insulina11.
La existencia de Ob-Rb en hígado, músculo esquelético y tejido adiposo indica que la leptina actúa en estos tejidos y, posiblemente, afecta la respuesta a insulina. Sin embargo, aún es insuficiente la evidencia que aclare la función fisiológica de estos receptores. Los estudios in vivo muestran que la administración central o periférica de leptina incrementa la sensibilidad a la insulina y la utilización de glucosa. Otros autores han mostrado que la leptina antagoniza el efecto de insulina en el hepatocito, por disminución de la fosforilación del insulin receptor substrate-1 (sustrato-1 del receptor de insulina, IRS-1). En el músculo esquelético, los resultados in vitro son contradictorios, pues mientras en algunos se observó que la leptina modula la acción de insulina, en otros no se determinaron efectos. Se especula que el rol in vivo de la leptina sobre el metabolismo de la glucosa está mediado, principalmente, por acciones centrales12.
En el tejido adiposo se destaca la actividad inhibitoria de la leptina sobre la lipogénesis y el incremento de la β-oxidación, la adipoapoptosis y los niveles de la proteína desacoplante 1 (UCP-1), que desacopla la cadena transportadora de electrones mitocondriales y disipa como calor la energía del gradiente electroquímico. La acción es directa autoparacrina y, además, mediada indirectamente por eferencias del SNA13.
La producción (síntesis y secreción) controlada y combinada de varias adipocinas y hormonas de los islotes pancreáticos (insulina y glucagón) establece una compleja red de moduladores del balance energético del organismo que controlan varios procesos tales como: el metabolismo de los lípidos (lipoproteína lipasa; LPL), el sistema inmune (TNFα, IL-1, IL-6, etc.) y la homeostasis vascular (por el factor de crecimiento endotelial vascular [VEGF], el fibrinógeno, PAI-1, angiotensina II, entre los más importantes).
La producción de múltiples adipocitocinas (TNF-α e IL-6, entre otras)14,15 favorece en las personas con obesidad hipertrófica el aumento de la la expresión local de leptina y varias adipocinas proinflamatorias tales como IL-8, IL-18 y MCP-116. El TNF-α también induce insulinorresistencia, por lo cual, en las personas obesas se halla una relación inversa entre sus niveles circulantes y la sensibilidad a la insulina. La reducción de peso corporal disminuye la concentración plasmática de TNF-α y mejora la sensibilidad a la insulina17. El TNF-α actúa a diferentes niveles para disminuir la sensibilidad a la insulina: reduce la activación del receptor para insulina, la fosforilación del IRS-1, así como la síntesis y la translocación del transportador de glucosa-4 (GLUT4)18. También inhibe la actividad del promotor de adiponectina (adipocina insulinosensibilizadora) y su concentración en plasma se correlaciona con cambios en la sensibilidad a la insulina19. Se ha observado que cuando los ratones KO para adiponectina se alimentan con una dieta rica en hidratos de carbono desarrollan insulinorresistencia severa20. El PAI-1 se expresa en adipocitos y su concentración plasmática se vincula directamente con la masa del TAB, tanto en humanos como en modelos animales21.
La leptina juega un papel clave en la ingesta de alimentos y la regulación del gasto energético mediante el control de la saciedad y del peso corporal a nivel hipotalámico22. Su acción está mediada por la activación de Ob-Rb, con alta expresión en el hipotálamo, cerebelo y otros tejidos tales como el páncreas y el hígado23. La activación de Ob-Rb induce la ruta JANUSK/STAT, con importante influencia en la modulación de la transcripción de la proteína24. La leptina inhibe la lipogénesis y aumenta la β-oxidación, la apoptosis de los adipocitos y la UCP-1, directamente por un mecanismo autocrino e indirectamente a través de vías eferentes del SNA25.
La leptina inhibe la expresión génica y la secreción de insulina, lo que afecta a la homeostasis de glucosa mediante la combinación con un efecto directo en el hipotálamo26. Debido a que la insulina estimula la liberación de leptina por los adipocitos, de esta interacción se forma un círculo de retroalimentación entre las células β y el TAB.
La leptina también inhibe la secreción de glucagón, por acción directa sobre las células α⋅ Los ratones con una alteración de la señalización de leptina desarrollan hiperglucagonemia27, mientras que la inducción de hiperleptinemia reduce los altos niveles de glucagón en ratones diabéticos. La insulina y el glucagón tienen efectos opuestos sobre la deposición/eliminación de sustratos metabólicos. La insulina promueve el almacenamiento, en cambio, el glucagón aumenta los niveles de glucosa en la sangre a través de la inducción de la producción de glucosa y su movilización desde el hígado28. La leptina antagoniza los efectos de glucagón en el hígado29, estimula la hidrólisis de triglicéridos y la oxidación de AGL, disminuyendo de ese modo tanto sus niveles circulantes como el almacenamiento tisular30, efecto protector contra el daño tisular inducido por la deposición de lípidos31. En conjunto, estos datos indican que el eje adipoinsular desempeña un papel regulador clave en el equilibrio energético en el que la insulina, el glucagón y la leptina son los mensajeros periféricos activos. Por lo tanto, la disfunción de este eje puede facilitar el desarrollo de la obesidad y la DMT232.
En las personas obesas se ha observado leptinorresistencia y se han propuesto varios mecanismos hipotalámicos para explicar este proceso22. La homeostasis de la energía requiere un equilibrio preciso entre la utilización de sustratos metabólicos (principalmente glucosa y AGL) por los órganos periféricos (hígado, músculo, TAB y tejido adiposo marrón), y de la disposición exógena (dieta) o endógena (hígado, tejido adiposo y riñón). El principal objetivo fisiológico es mantener un nivel de glucosa que garantice la provisión al cerebro. Este complejo sistema regula, involucrando una estructura «pivot» como el hipotálamo, la actividad metabólica de la corteza corticoadrenal, el TAB, el páncreas endocrino y, por ello, el hígado y el músculo (entre otros tejidos)33. Una visión integrada de este eje se representa en la figura 1.
Ácidos grasos libres en obesidad y diabetes: estimulación; (-): inhibición. Fuente: Adaptado de Pagano et al. Neuroendocrinology. 2017;104:347-363.")
Los AGL se encuentran elevados en la mayoría de las personas obesas y con DMT2 y se considera que intervienen en el fenómeno de insulinorresistencia, tanto a nivel periférico (muscular), como en el hígado. Asimismo, a la elevación de los AGL se la vincula con otros factores de riesgo cardiovascular como las dislipidemias, la hipertensión arterial y la hiperuricemia. También en la actualidad se la relaciona con la declinación de la célula β.
El criterio más reciente es que existe un fallo en la supresión de la lipólisis que fisiológicamente se debería producir por actividad de la insulina. Se ha hallado una relación estrecha entre la elevación de los AGL plasmáticos, el aumento de la grasa intrafibrilar muscular y la insulinorresistencia.
Randle, de Cambridge, marcó un hito cuando en 1963, bajo el concepto del «ciclo glucosa-ácidos grasos» reunió fisiológicamente a los metabolismos glucídico y lipídico e introdujo además la idea de que la anormalidad de los AGL interviene en la etiopatogenia de la DMT2. Estimó que se ubicaba en el centro de la regulación del uso de la energía corporal, ya que el aumento de la oxidación de las grasas en ayunas (que se acompaña de un bajo nivel de insulina) sirve para aportar energía, pero también para disminuir la captación de glucosa por el músculo y así preservar la glucemia, especialmente para proveer de combustibles al cerebro (el gran consumidor de glucosa). En cambio, cuando se absorben nutrientes, la insulina se eleva y estimula el depósito de glucosa en el glucógeno e inhibe la lipólisis por lo que, de esta manera, promueve el depósito de fuentes de energía34.
Además, su elegante modelo constituyó por largo tiempo la brillante hipótesis sobre la manera por la que la lipólisis acentúa la inhibición de la captación muscular de glucosa por las grasas. De esta manera explicó, aunque fuera parcialmente, como las grasas disminuyen la sensibilidad a la insulina (o promueven insulinorresistencia) en la fisiopatología de la DMT2. En letra pequeña escribió que «la insensibilidad a la insulina se podría deber a una disminución de la respuesta del transportador de glucosa a la insulina o a la inhibición de la fosforilación de la glucosa». Consideró que el bloqueo de la fosforilación y las enzimas hace que se acumule glucosa libre (en la célula), lo que restringe, finalmente, el ingreso de la glucosa (desde la circulación) a través del transportador de glucosa (GLUT4).
También admitió que «no es posible responder si la aceleración de la lipólisis es el único mecanismo involucrado en la disminución de la sensibilidad a la insulina». Sin embargo, se tardó más de 40 años en probar que había errores cardinales en parte de estos conceptos.
Sus hipótesis tuvieron una base experimental y en lo esencial estudió la transferencia de AG a la albúmina del plasma en el tejido adiposo y la lipólisis en el diafragma y cardiomiocitos de rata (agregado de AG al medio de perfusión). Su particular propuesta comenzó a revisarse tiempo después, porque si bien tenían una prolija investigación, la mayor parte se había realizado con tejidos in vitro o en roedores, e incluso Shonfeld y Kipnis, corto tiempo después, no lograron reproducir los resultados de Randle en diafragma de rata35.
Luego de iniciar incipientes trabajos sobre la participación de las grasas en el metabolismo de la glucosa36, pasaron 20 años hasta que Ferraninni y DeFronzo aportaron evidencias que comenzaron a clarificar la actividad de los AG en humanos. Investigaron el efecto de la elevación fisiológica aguda de AG sobre la producción y captación de glucosa en sujetos con tolerancia normal a la glucosa bajo 3 situaciones de ensayo controlado (estudios con el método del «clamp» o pinza glucémica insulinémica) en situaciones que imitasen el estado normal en ayunas y el de DMT237.
La elevación fisiológica aguda de los AGL plasmáticos en el músculo frenó la utilización de glucosa estimulada por insulina pues hay «competición de sustratos entre la glucosa y los AGL». De ahí que la insulina mejora aún más la utilización de glucosa cuando inhibe la lipólisis y caen los AGL del plasma.
En contraste, ante un nivel bajo de insulina con glucemia moderadamente elevada (como en la DMT2) la infusión de lípidos no afectó significativamente a la utilización de la glucosa. Se consideró que tal vez ante la falta de la hormona, la mayor parte de la glucosa se utiliza en tejidos independientes de insulina (principalmente células nerviosas y eritrocitos) y existe una pobre intervención del tejido muscular.
Ferraninni y DeFronzo admitieron en sus primeros trabajos que se requería de más investigaciones para definir los mecanismos, pero que los resultados eran compatibles con la forma de operar del ciclo glucosa-ácidos grasos, in vivo. Por un lado, la glucosa cuando estimula la secreción de insulina, por su efecto antilipolítico reduce los AGL. Por el otro, cuando existe un aumento de la demanda, los AGL compiten efectivamente con la glucosa, primordialmente en el músculo esquelético y en el miocárdico. Se interpretó en aquel momento, que habría solo una limitada competencia de sustratos responsable por la resistencia a la insulina en la DMT2 y es poco probable que el aumento del aporte de AGL influya sobre el metabolismo de la glucosa, ya alterado cuando la secreción de insulina es deficiente. En cambio, la lipólisis acelerada aporta sustratos que estimulan la gluconeogénesis (formación de glucógeno a partir de sustancias que no son glúcidos) que, a su vez, contribuye, por aumento de la producción hepática de glucosa, a la hiperglucemia en sujetos con déficit de insulina.
En la primera mitad de la década del 80, se consideró que existían aceptables evidencias de la relación entre la hiperglucemia, la alteración para utilizar la glucosa en el músculo y la dificultad para frenar la producción endógena de glucosa hepática. En cambio, a pesar de que desde 1953 ya se contaba con resultados en diabetes experimental, pasó un largo tiempo para que se conocieran las características del efecto antilipolítico de la insulina sobre el tejido adiposo y sus consecuencias en la diabetes38,39. En 1985 Fraze y Reaven et al., para evitar confusiones y en contraste con estudios previos que determinaron los AGL plasmáticos solo como parte de una prueba de tolerancia a la glucosa oral durante la mañana, midieron y compararon la glucosa, la insulina y los AGL plasmáticos en 15 diabéticos no obesos y en 15 controles con tolerancia a la glucosa normal, a quienes se les administró comidas regulares (08:00, 12:00 y 18:00 h)40.
Las glucemias, las insulinemias y los AGL (con excepción de las 11:00 y 12:00 h) fueron significativamente más elevadas en los pacientes con DMT2 que en los sujetos control.
Este estudio fue el primero que demostró que las concentraciones de AGL:
- 1.
Son más elevadas en las personas con DMT2 ante comidas normales,
- 2.
tienen un defecto metabólico proporcional a la severidad de la hiperglucemia y
- 3.
aumentan a pesar de la presencia de niveles elevados de insulina.
Fraze y Reaven agregaron que «así como los pacientes con DMT2 presentan una alteración en la captación de glucosa estimulada por la insulina, tienen además un defecto en los niveles de AGL», aunque no definieron si la causa era una lipólisis acelerada o la disminución de la reesterificación de los AGL.
Pocos años después, Swislocki junto al equipo de Reaven profundizaron los estudios y analizaron bajo condiciones controladas de glucemia e insulinemias (clamp), las curvas «dosis-respuesta» en personas con DMT2 y en controles (con tolerancia a la glucosa normal) y se determinaron las concentraciones de AGL del plasma. Esta vez, se mantuvieron niveles similares de insulina en los grupos de estudio para evitar que los cambios en los AGL se debieran a diferencias en las insulinemias41. Se observó que, aún en caso de que la insulina preserve su acción supresora sobre los AGL, estos fueron más altos en la DMT2 que en los controles, por lo cual se concluyó que, si bien los niveles de insulina se elevaron en los diabéticos de tipo 2, la hormona no fue capaz de controlar los AGL.
McGarry con enorme audacia revisó desde otro ángulo la fisiopatología de la DMT2 e inició su labor con el interrogante «¿es posible que nuestra percepción histórica del disbalance metabólico primario en la diabetes haya puesto el énfasis en aspectos erróneos? Tal vez los 2descubrimientos fundamentales influyeron decisivamente en nuestra forma de pensar»42.
Refiere la leyenda que, en 1889, avisaron a Oskar Minkowski de que una cantidad inusual de moscas sobrevolaban la orina de sus perros pancreatectomizados. Se dice que probó el gusto de la orina y se asombró por su sabor dulce. De esta astuta observación estableció que había alguna sustancia del páncreas que manejaba los azúcares del cuerpo. En su ausencia, aparecía la diabetes. Desde entonces se centró la visión de la diabetes en el metabolismo glucídico y los conceptos giraron a su alrededor, incluso cuando Banting y Best descubrieron la insulina. Pero McGarry se preguntó ¿y si Minkowski no hubiese tenido el sentido del gusto (ageusia)? Seguramente habría percibido el «olor a acetona» y concluido que la remoción del páncreas provoca alteraciones en el metabolismo de las grasas. Es altamente probable que luego Banting considerara que el rol preeminente de la insulina era controlar el metabolismo de los lípidos. Y el devenir habría sido distinto.
En 1994 Reaven dictó la conferencia Claude Bernard en la European Association for Study of Diabetes y no fue menos ingenioso con su título: «El cuarto mosquetero, de Alejandro Dumas a Claude Bernard».
El tejido adiposo en condiciones fisiológicas es altamente sensible a la insulina y pequeños aumentos en las concentraciones de insulina provocan la supresión marcada de los AGL. Sin embargo, los AGL no se inhiben en los diabéticos de tipo 2 debido a que el tejido adiposo es resistente a la actividad antilipolítica de la insulina y su nivel supera las concentraciones plasmáticas de las personas con tolerancia normal a la glucosa (regulación «disfuncional» de los AGL en la DMT2). Esto produce 3efectos nocivos:
- 1.
La disminución de la depuración o extracción de la glucosa circulante;
- 2.
la alteración de la supresión de la producción hepática de glucosa (glucosa endógena);
- 3.
la declinación posterior de la secreción de insulina.
En ella incluyó y enfatizó la participación del tejido adiposo en la DMT2. Señaló que, junto al músculo, el hígado y la célula β, el tejido adiposo juega un papel clave en la patogénesis de la enfermedad. Ya que DeFronzo había denominado «el triunvirato» (hígado, músculo, célula β), él puso un cuarto componente como «nuevo mosquetero» (el tejido adiposo)43. Luego, el mismo DeFronzo sumó la alteración de las grasas y comenzó a hablar del paso del triunvirato al «cuarteto disarmónico».
También los trabajos de Boden confirmaron en humanos la disminución en la oxidación de hidratos de carbono cuando hay oxidación de grasas, con una reducción del 40 al 55% de la captación muscular de glucosa (esto tanto en sujetos control como en diabéticos)44,45. Desde que Randle consideró que el aumento de la oxidación de lípidos era la causa de la disminución de la captación muscular de glucosa, se acumularon evidencias que reunieron la elevación de los AGL con el desarrollo de resistencia a la insulina en el músculo, pero sin duda fue a partir de los trabajos de Dressner y Shulman cuando cambió radicalmente el concepto sobre la insulinorresistencia muscular. El grupo de Yale con el aporte de nuevas técnicas aplicó la combinación de espectroscopia de resonancia magnética nuclear, de biopsia muscular en 14 voluntarios sanos y de una enorme lucidez.
Sus resultados mostraron que el aumento y manutención de AGL por 5 h durante el «clamp» euglucémico hiperinsulinémico inhibió entre el 50 y 60% la captación corporal de glucosa y provocó una disminución de la síntesis del glucógeno, al comparar con el grupo control, en el que no se elevaron los AGL. Esto se acompañó con una bajada casi del 90% en la actividad de la glucosa 6 fosfato muscular, lo que planteó la posibilidad de que se inhibiesen una de estas 2 vías: o del trasportador o de la fosforilación celular de glucosa. Cuando los estudios se profundizaron, se halló la disminución de la glucosa intracelular y se interpretó que la menor captación muscular se debe a la inhibición del transportador de glucosa.
En una investigación posterior, lograron determinar que el estímulo con insulina y la infusión de glicerol producen en el músculo un aumento de la enzima fosfoinositol 3 cinasa, cuya actividad se vincula al transportador de glucosa. En cambio, la reacción enzimática no se produjo cuando la infusión se realizó solo con lípidos.
De allí, que las evidencias muestran que el aumento de los AGL altera la señal de insulina. La elevación de AG dentro de la célula muscular tiene un efecto depresor sobre el IRS-1 asociado a la actividad de fosfoinositol 3 cinasa46.
En la actualidad se conoce que para activar el receptor de insulina existe un proceso de fosforilación en tirosina. En 2002 el mismo grupo, luego de un trabajo experimental en ratas, propuso la hipótesis de que el aumento de la concentración de AGL plasmáticos provoca un incremento en los niveles intracelulares de AG acyl-CoA, diacilglicerol y ceramidas que activan a la proteína cinasa C (theta), lo que aumenta la fosforilación del IRS-1 en serina. Esto disminuye la fosforilación en tirosina del IRS-1, baja su activación asociada a fosfoinositol 3-cinasa y «apaga» la actividad del transportador de insulina47. Así disminuye la sensibilidad (actividad) de la insulina a nivel muscular.
Si bien la investigación básica mostró diversos e interesantes aspectos de la importancia de los AGL como factor que influye en el desarrollo de la DM, desde el punto de vista epidemiológico no se han realizado estudios prospectivos en escala que den el mismo sustento, como el que se observó en la experimentación. Uno de las primeras publicaciones fue del grupo de Ravussin en sus clásicos estudios, donde examinó anualmente a 190 indios pima no diabéticos por prueba de tolerancia oral a la glucosa, «clamp» euglucémico hiperinsulinémico y células abdominales, en la que estudiaron lipólisis in vitro. En un tiempo medio de 4 años, 47 sujetos desarrollaron una DMT2 y los factores predictivos fueron: el tamaño de los adipocitos y el nivel de los AGL en ayunas48. Otro trabajo destacado fue el Paris Prospective Study, que es el primero que demostró la importancia de los AGL, pero esta vez en 4.089 caucásicos y enfatizó que los niveles de AGL predijeron el paso de tolerancia normal a tolerancia alterada a la glucosa y también fueron predictores del paso de esta a DMT2. Además, el 60% de las personas con tolerancia alterada a la glucosa tuvieron AGL en ayunas que se ubicaron en el tertilo superior49. También el estudio Atherosclerosis Risk in Communities (ARIC) documentó el aumento del riesgo relativo de desarrollar DM en sujetos con AGL elevados50.
En sentido inverso, se ubicó el estudio Ely que se hizo en Cambridge, como análisis de cohortes prospectivo de 481 mujeres y 345 hombres en quienes se realizó en una prueba de tolerancia a la glucosa oral y la determinación de AGL, que se repitió en un seguimiento a 4,5 años. Los datos no mostraron una relación longitudinal entre los AGL basales y la alteración en la tolerancia a la glucosa u otras características del síndrome metabólico, durante el seguimiento. En contraste, se observó una fuerte asociación en análisis de corte entre los AGL y la tolerancia a la glucosa o rasgos del síndrome metabólico tanto en la base como en el seguimiento. Hallaron diferencias significativas en los niveles de AGL y las mediciones de supresión de AGL en sujetos con criterios de síndrome metabólico y en aquellos con tolerancia normal a la glucosa. Por estas razones, los autores consideraron que los niveles de AGL plasmáticos cambian como consecuencia del síndrome metabólico y que no tiene sustento la intervención de los AGL como causa de la DM o del síndrome metabólico51.
La porfía sobre la influencia de los AGL sobre el desarrollo de resistencia a la insulina y la DMT2, a qué nivel o circunstancias actúan y cómo lo hacen, permanece en acalorada discusión. Para Boden y Laakso las evidencias siguen acumulándose en el sentido de que la elevación crónica de los AGL representa un evento temprano en la patogénesis de la DMT2. Los AGL inducirían resistencia a la insulina y consideran que se trata de una respuesta adaptativa beneficiosa, particularmente en el ayuno y el embarazo. Sin embargo, ante la sobrealimentación y el sedentarismo, las grasas se atesoran en el tejido adiposo visceral y subcutáneo y en la medida que el cúmulo de grasas aumenta, se elevan los niveles de AGL y esto conduce a la insulinorresistencia. Por supuesto que el exceso de algunos productos o el déficit de otros (como adiponectina) van a acelerar el fenómeno de insulinorresistencia. Para amortiguar el efecto de esta situación aumenta la secreción de insulina. En personas con predisposición génica por la DM, la célula β va a claudicar y se va a desarrollar la enfermedad clínica52.
Sin embargo, esta es una propuesta demasiado simple de la relación entre los niveles de AGL y el fallo de la célula β y es probable que sea un efecto que se podría considerar de corto o mediano plazo. No puede obviarse la exposición crónica a niveles elevados de grasas que resulta tóxica a la célula β y que afecta su función y su sobrevida. De allí que el fallo β-celular vaya más allá del efecto o las consecuencias de las insulinorresistencia hepática y muscular53.
La grasa abdominal perivisceral: la hipótesis portal de resistencia a la insulinaEs probable que desde la impresionante clasificación clínica de Lanceraux en diabetes «magra» y diabetes «grasa» (Diabète maigre et diabète gras) que se ubica entre 1879 y 1899 (Traité des maladies du foie et du pancréas y Diabète pancréatique) se reconoce el vínculo entre la obesidad y la diabetes. Sin embargo, se plantea un cierto grado de incertidumbre cuando se discute por qué hay personas obesas sin insulinorresistencia y sujetos conIMC normal, con manifestaciones de resistencia a la insulina54,55. Las primeras explicaciones consideraron que la adiposidad de tipo central o abdominal («androide») era la que establecía la relación entre obesidad, DMT2 y enfermedad cardiovascular56 y luego, que el índice cintura/cadera aparecía como un recurso clínico subrogado para distinguir a los individuos con y sin riesgo de enfermedad cardiometabólica. Si bien Kissebah57 publicó en primer término, fue el grupo de Gotemburgo en Suecia, el que, a través de un prolongado estudio en el que se siguió durante 13,5 años a hombres y durante 12 años a mujeres, demostró que el índice cintura/cadera era un fuerte predictor de desarrollo futuro de diabetes, angina pectoris, infarto de miocardio, accidente cerebral vascular y muerte, independiente del IMC58,59.
Con el transcurso del tiempo y con los adelantos técnicos fue creciendo el conocimiento sobre la relación entre la insulinorresistencia, la DMT2, la enfermedad cardiovascular y el tejido adiposo corporal total, el abdominal perivisceral y el depósito subcutáneo60–63.
Para un grupo de investigadores es el activo depósito abdominal perivisceral de grasa el que se relaciona con la insulinorresistencia, la DMT2 y la enfermedad cardiovascular, a pesar de su menor tamaño con respecto a la adiposidad subcutánea y general.
La hipótesis portal considera, en definitiva, que la distribución del tejido adiposo es un predictor importante e independiente de insulinorresistencia, DMT2 y enfermedad cardiovascular y que el exceso de grasa abdominal —en particular omental y mesentérica (y algo en retroperitoneo) de alto dinamismo (o sea con intenso recambio)— presenta una lipólisis que produce una plétora de AGL, que por vía porta llegan al hígado.
Fue muy intensa la influencia del trabajo de Jensen et al., quienes en 1989 midieron el recambio de palmitato como marcador de lipólisis sistémica en 10 mujeres con obesidad troncal («alta»), 9 con obesidad femoroglútea («baja») y 8 no obesas y hallaron que las mujeres con obesidad alta tuvieron un mayor recambio de palmitato, aunque una menor respuesta lipolítica a epinefrina que las de obesidad baja y las de normopeso. Además, todas las obesas mostraron una alteración en la supresión del recambio de AGL en respuesta a la insulina, cuando se compararon con las no obesas. De allí que concluyó que las distintas respuestas metabólicas correspondían a diferentes tipo de obesidad por heterogeneidad en los adipocitos64.
Hubo investigaciones epidemiológicas que mostraron la importancia de la obesidad central. El Estudio de las Enfermeras (Nurses’ health study) fue una investigación que demostró una fuerte relación entre el IMC como parámetro de obesidad y el riesgo de desarrollar DMT2, entre las mujeres65. De la misma manera, en el Estudio de los Profesionales (Male health professionals) se afirmó que la relación entre obesidad y DM es causal y además que la circunferencia de cintura es mejor indicador que el índice cintura/cadera para determinar la relación entre la adiposidad abdominal y el riesgo de DM66. El Estudio de los japoneses americanos incluyó a sujetos de segunda y tercera generación sin DM y, luego de 10 años, el principal predictor de incidencia de DM fue el área de grasa intraabdominal. Se concluyó que la obesidad abdominal es un factor de riesgo de DMT2 independiente de la glucemia en ayunas, la secreción de insulina, la adiposidad regional y de la historia familiar de DMT267.
Bergman propuso que la insulinorresistencia del hígado deriva de un aumento relativo en la liberación de AGL desde el depósito omental al hígado. Las causas presuntas serían68:
- 1.
Un alto depósito de lípidos en el tejido omental,
- 2.
una resistencia a la insulina severa del depósito central de grasa y
- 3.
una posible intervención del sistema nervioso central en la regulación de la lipólisis.
Se considera que, cuando hay sobrecarga hepática de AGL, se produce un racimo de fenómenos69:
- 1.
Aumenta la producción de VLDL cargadas de triglicéridos y de apolipoproteína B (aterogénicas);
- 2.
se incrementa la neoglucogénesis hepática y
- 3.
disminuye fuertemente la capacidad de aclaramiento de la insulina, que llega también por vía porta.
Se acepta así que los AGL tienen un importante control sobre la producción hepática de glucosa. La exposición a una cantidad exagerada de AGL provocaría una insulinorresistencia hepática que el resto del organismo intacto intenta compensar. En un principio solo se consideró que lo haría con mayor cantidad de insulina que, en definitiva, tendría 2orígenes:
- 1.
Mayor secreción de insulina, ya que los propios AGL —en niveles moderadamente elevados— sensibilizan a la célula β que produce y libera más hormona, y
- 2.
menor degradación hepática, lo que se refleja en una hiperinsulinemia «compensadora»70.
El grupo de Bergman efectuó un trabajo con perros en los que se indujo una obesidad central y a los que se les colocaron catéteres en la arteria mesentérica superior para insulinizar discretamente el lecho del tejido adiposo visceral. La infusión omental no bajó a los AGL sistémicos, posiblemente porque la insulinorresistencia de la grasa omental produjo un requerimiento que cuadruplicó los niveles fisiológicos, que son antilipolíticos en el resto del cuerpo. Posteriormente, agregó una inferencia interesante: es posible que la disminución del aclaramiento o eliminación de la insulina por el hígado ayude para que la hiperinsulinemia mantenga la glucosa en niveles normales con menor esfuerzo para la célula β que, de por sí, tiene una sobreproducción de insulina forzada por la presencia de insulinorresistencia. Estima que tal vez este es el primer mecanismo de defensa para preservar la célula β.
Kahn et al., que han estudiado la influencia de la resistencia a la insulina sobre la función de la célula β (curva de relación entre el índice de sensibilidad a la insulina y el pico agudo de secreción de insulina), afirman que el envejecimiento deteriora la función β-celular, pero uno de los factores que inciden sobre este decaimiento funcional es el cúmulo de la grasa abdominal que ocurre con el pasar de los años71.
En la actualidad se admite que la respuesta del organismo a la adiposidad central es compleja y que, en el intento de mantener el equilibrio, intervendrían también las enterohormonas como el glucagon like peptide (GLP-1) y el glucagón pancreático, que se sumarían a la insulina para el manejo de la producción hepática de glucosa.
Por otra parte, se especuló que si una gran cantidad de AGL alcanza a tejidos dependientes de insulina (como el músculo al que llegan por medio de VLDL o a partir del tejido adiposo subcutáneo) disminuye la sensibilidad a la hormona a través del efecto de Randle.
De allí que, para otro grupo de investigadores, es el tejido adiposo subcutáneo el que se relaciona con la resistencia a la insulina o, al menos, consideran que tanto la adiposidad subcutánea como la perivisceral influyen por igual72–74.
Las técnicas de imágenes como la tomografía computada y la resonancia magnética a la altura de lumbar 4-5 han permitido comparar las áreas visceral y subcutánea del tejido adiposo. Un programa que examinó el abdomen en franjas facilitó el cálculo del volumen de la adiposidad abdominal, subcutánea y total. Más aún, se subdividió la masa de tejido adiposo y se estimó que dentro del compartimiento abdominal existe un área retroperitoneal que presumiblemente drena a la circulación general y otro que se vuelca a la circulación portal75 y que el depósito subcutáneo posee una capa profunda que se relaciona con el fenómeno de insulinorresistencia y una superficial, sin ningún clase de vínculo con ella39.
En sentido estricto, la mayor parte de las propuestas que estiman que en la grasa abdominal se ubica el punto de partida de las alteraciones glucosa/insulina que finalmente culminan en la insulinorresistencia tienen argumentos indirectos. Se discute que sea el factor causal y se considera más bien como otro de los mecanismos potenciales que reúnen a la obesidad central y a la DMT276,77. No se deja de reconocer que el tejido adiposo subcutáneo posee una masa adiposa considerablemente mayor que, de alguna manera, influiría sobre la sensibilidad a la insulina por el volumen de AGL que libera a la circulación general, lo que se considera un fenómeno de insulinorresistencia de mecanismo no portal78,79.
Por otro lado, se ha demostrado que la reducción selectiva de la adiposidad visceral a través de cuidados alimentarios y ejercicio se acompaña de una mejora significativa del metabolismo intermedio y de la reducción de los factores de riesgo de enfermedad cardiovascular80,81. Es también llamativo el trabajo de Gabriely et al., quienes estudiaron la actividad de la insulina, la tolerancia a la glucosa y la expresión de los péptidos derivados, después de eliminar el tejido adiposo visceral (18% de la grasa total) en ratas ancianas Brown Norway/F344 (BNF) y Zucker obesas diabéticas (ZDF). La acción hepática y periférica de la insulina mejoró y fue comparable a la actividad que se observa en ratas jóvenes BNF. Se evitó la disminución progresiva de la actividad de insulina y retardó el inicio de la DM y, aunque no modificó los niveles de AGL, sí descendió entre 2 y 3 veces la expresión de adipocinas como el TNF-α y la leptina del tejido adiposo subcutáneo, en el grupo de ratas ZDF. Para los autores, los resultados demuestran en forma experimental que existe una relación causal entre la grasa visceral y la resistencia a la insulina82.
En realidad, se requiere aún de información sobre los determinantes genéticos y epigenéticos de la topografía y la biología molecular del tejido adiposo visceral. De allí que la fuerza de la hipótesis portal se fuera limitando en la medida en que fueron creciendo los conceptos del depósito ectópico de grasas y del paradigma endocrino, en el que el adipocito, además de los AGL, secreta factores que influyen así sobre el metabolismo de otros tejidos distribuidos en el organismo83.
Limitación de la adipogénesisHirsch et al., a fines de los años 60, fueron de los primeros en considerar, a partir de sus experimentos, que la respuesta a la insulina es una función de la media del tamaño del adipocito. Mostraron que las células adiposas de obesos y no obesos metabolizaban igual la glucosa, los triglicéridos y el dióxido de carbono. En contraste, la respuesta del tejido adiposo a la insulina dependió del tamaño celular. Cuanto mayor era el adipocito, menor era la sensibilidad tisular a la insulina. Años después concluyeron que la hiperinsulinemia y la insulinorresistencia periférica de la obesidad se debían más a la hipertrofia adipocitaria que al aumento de grasa corporal total y a la ingesta alimentaria84,85. Es más, el agrandamiento adipocitario se relaciona con la insulinorresistencia sistémica con mayor fuerza que cualquier otro parámetro.
El concepto esencial es que si en los momentos de sobreabundancia de alimentos la masa grasa no se puede expandir a través de la proliferación y la diferenciación, los adipocitos se hipertrofian. Llegaría también a un cierto límite de la hipertrofia en la que tampoco puede guardar más grasas y así promovería el asiento de lípidos en otros tejidos y órganos.
No parece que el freno de la hipertrofia adipocitaria fuese un fallo, sino más bien una situación fisiológica que restringe el crecimiento «ilimitado» del tejido adiposo. Con el adipocito colmado, puede suceder que las grasas no ingresen o lo hagan, pero se movilicen con gran intensidad. Existen hoy, múltiples publicaciones e incluso algún estudio epidemiológico, como el de los indios pima, que apoyan el concepto de que la hipertrofia adipocitaria es consecuencia de la alteración de la proliferación y de la capacidad de diferenciación de los adipocitos y que también son factores precipitantes para el desarrollo de DMT286,87.
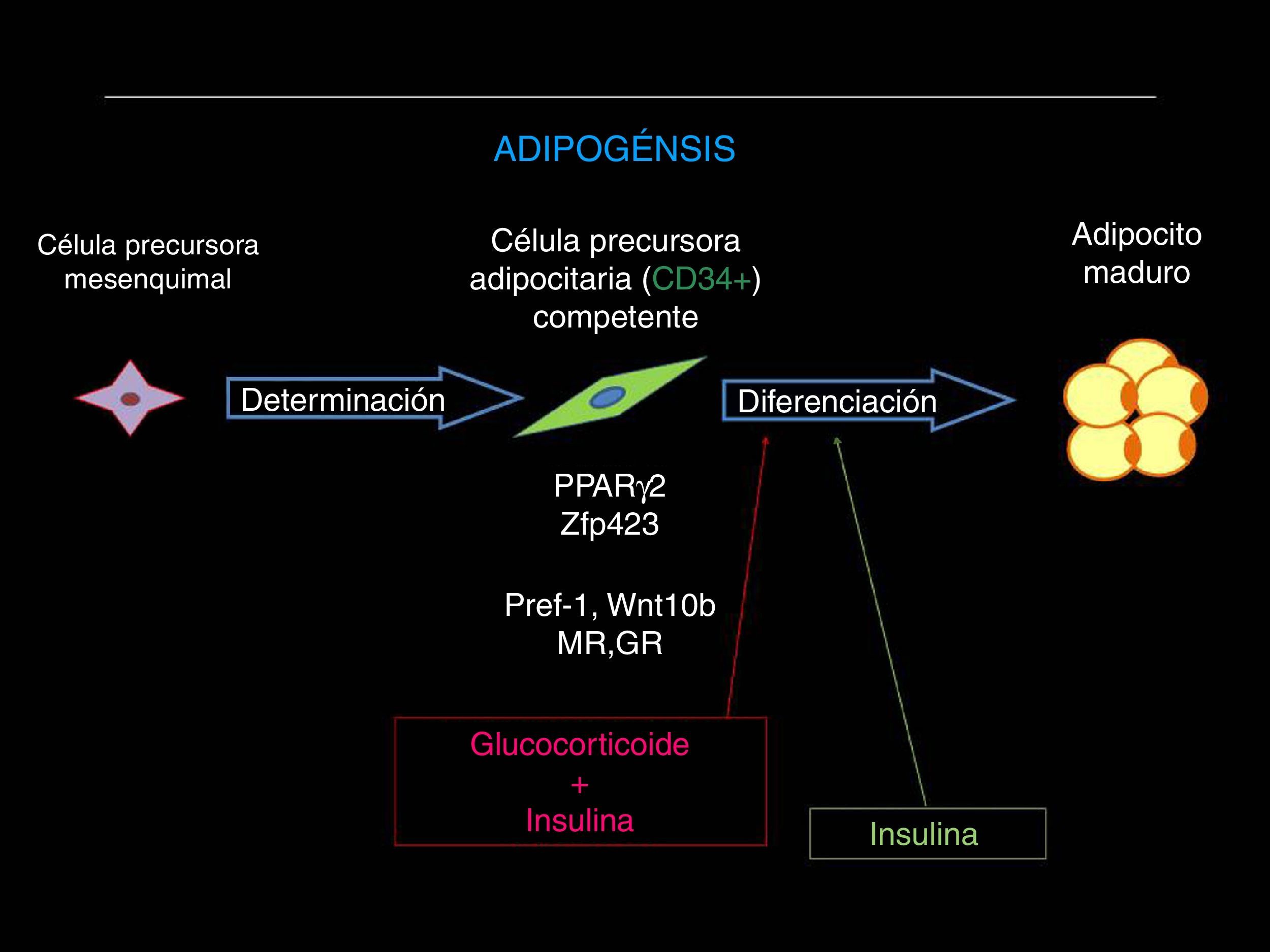
Al respecto, el proceso denominado adipogénesis incluye la proliferación desde una población de células pluripotenciales mesenquimales que, por expresar el antígeno CD34 (CD34+) se encuentran «determinadas» para diferenciarse en adipocito, y es la «competencia» celular la característica por la cual las células precursoras de adipocitos (CPA) adquirirán su capacidad para diferenciarse a adipocitos maduros (fig. 2). En este punto intervienen varios factores de transcripción (como el PPAR-γ2, SREB-1, C/EBP γ, β y δ) y otros elementos no transcripcionales influidos por señales extracelulares (adiponectina, leptina, TNF-α, etc.) en un complicado proceso que aún no se encuentra totalmente esclarecido88.

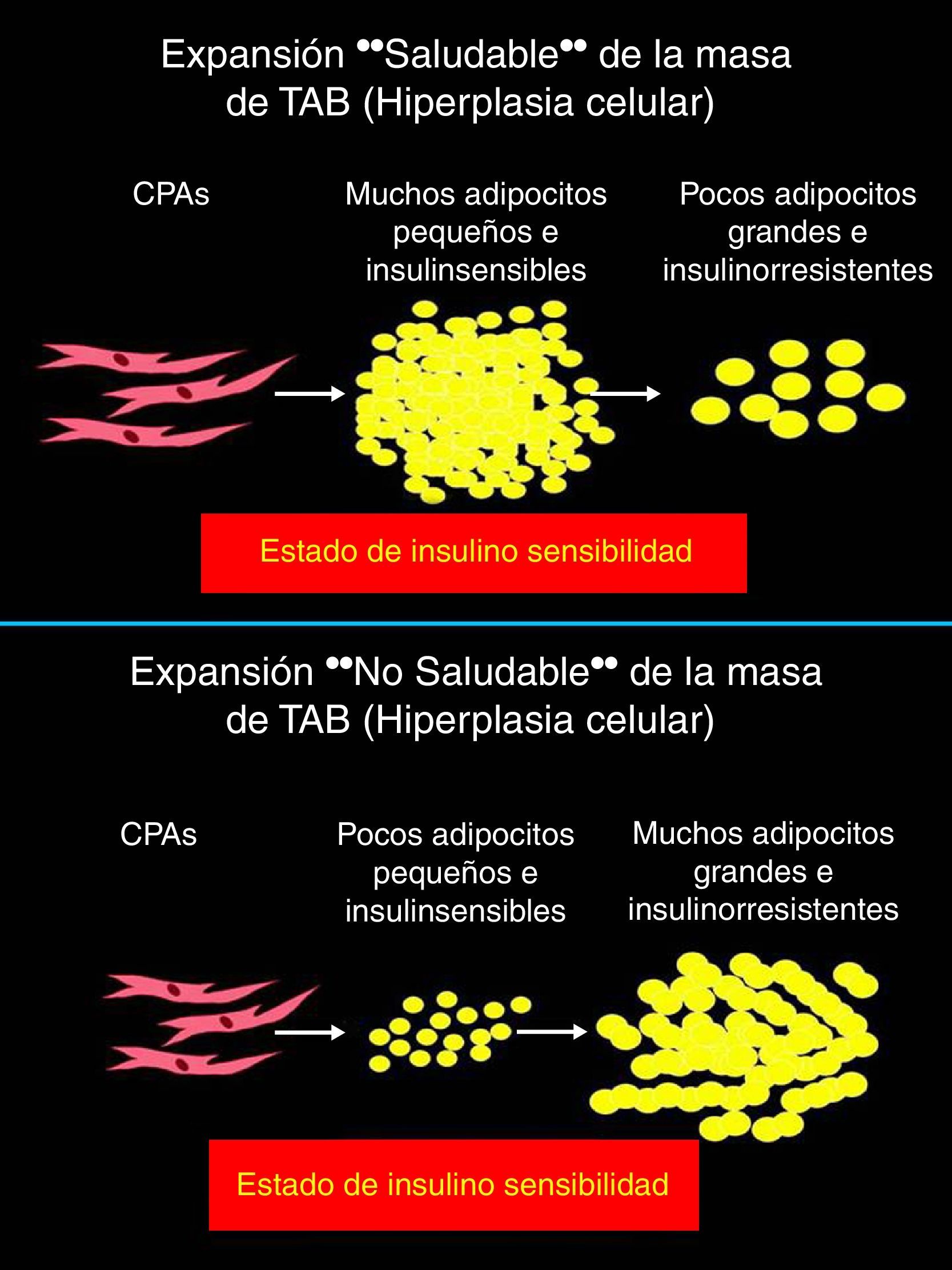
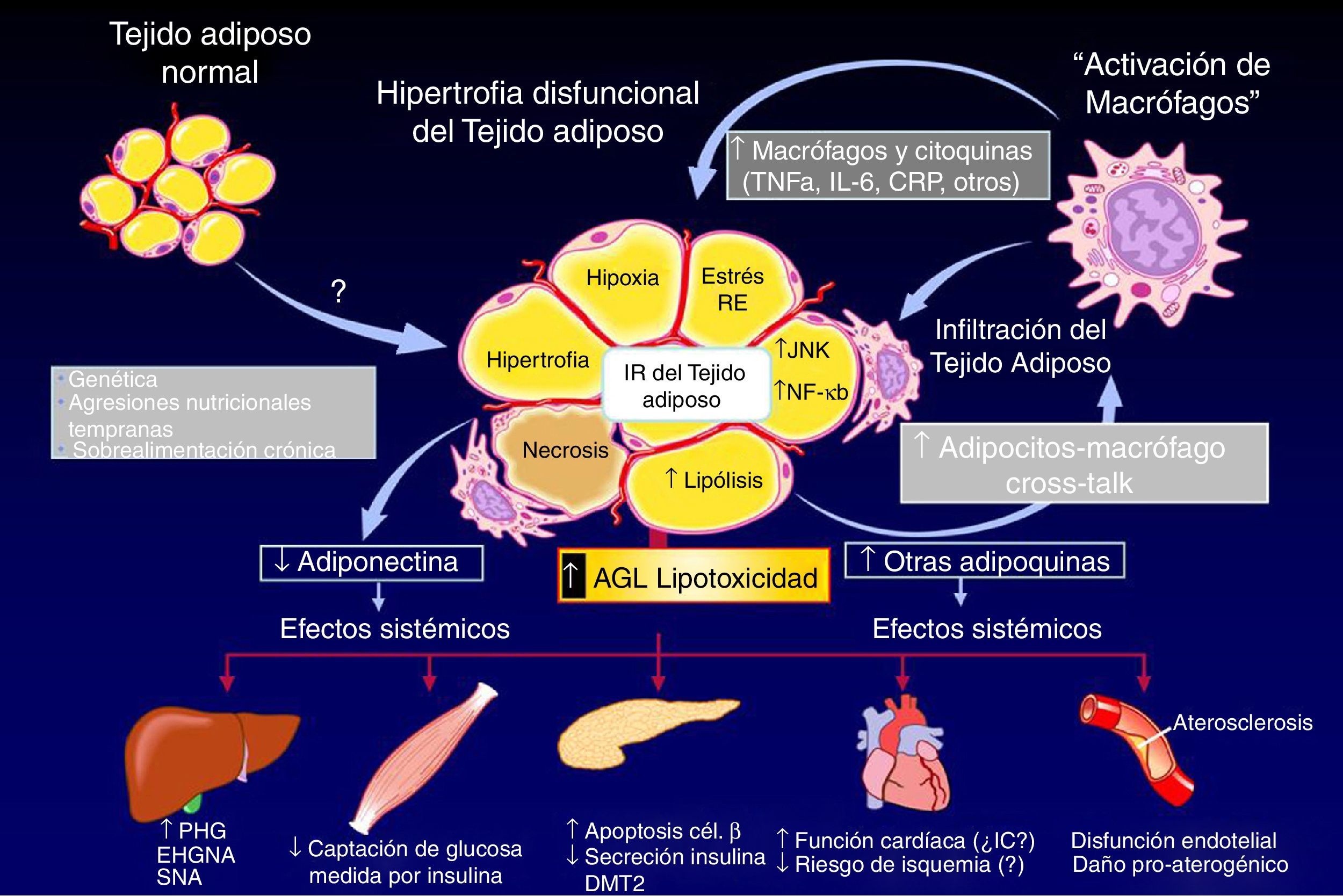
El proceso adipogénico, asociado al angiogénico local, forma parte de una importantísima actividad del TAB: su expansión en la masa celular. Ha sido claramente descifrado que la hipertrofia adipocitaria es una manera «no saludable» de expansión de la masa de TAB, que se diferencia de aquella «saludable» resultado de la neoadipogénesis (adipogénesis de novo) o expansión hiperplásica del TAB (fig. 3, panel superior). El adipocito hipertrófico es una fuente de producción excesiva de adipocinas proinflamatorias (fig. 3, panel inferior), célula que de una u otra manera es desconocida por el sistema inmunológico y resulta infiltrado por macrófagos (células productoras también de citocinas proinflamatorias), lo que exacerba el estado inflamatorio local (del TAB). Resultante de ello son los procesos sistémicos alterados y sus consecuencias sobre la salud del individuo (fig. 4). Hoy en día se sabe que hay varios componentes que son inductores de la hipertrofia adipocitaria, entre ellos el exceso endógeno crónico de glucocorticoide (similar al fenotipo síndrome de Cushing)89 o de andrógeno (similar al fenotipo síndrome de ovario poliquístico)90. Sin embargo, habría otros factores como los relacionados con los nutrientes ingeridos (dietas con alto contenido de hidratos de carbono, como sacarosa y fructosa, específicamente) que pueden resultar en la expansión «no saludable» de la masa de TAB. Este tipo de expansión del TAB llevó a clasificar los fenotipos resultantes como pertenecientes a los de «obesidad hipertrófica». Es importante resaltar que los factores endógenos primordiales en el proceso fisiológico de diferenciación de CPA son inicialmente los glucocorticoides más la insulina y, después, solo la insulina (ver fig. 2). Luego, un medio interno caracterizado por un prolongado exceso en la producción de estas señales origina una reducción en la población de CPA alojadas en la fracción estroma vascular local (TAB), un hecho asociado a una capacidad de diferenciación retardada91. La resultante es la generación de adipocitos maduros que rápidamente se hipertrofian; esa situación puede ser revertida corrigiendo la producción excesiva de aquellas señales91. Como antes se ha mencionado, el consumo excesivo de fructosa, a través de la ingesta de una dieta normocalórica, también induce una expansión hipertrófica de la masa de TAB (visceral), aunque de forma diferencial a la ya mencionada92. Es decir, si bien no se generan cambios en la población (número) de CPA en la fracción de estroma vascular del TAB, un disbalance de señales pro- (Zpf423 y PPARγ-2) y anti-(Pref-1 y Wnt-10b) adipogénicas, desplazado hacia la actividad de las primeras, resulta en la aceleración del proceso adipogénico en su etapa inicial al que sigue una hipertrofia celular92 debido a mecanismos que aún no se han esclarecido completamente.
 o a expensas del aumento del tamaño de los adipocitos maduros residentes en ese tejido: hipertrofia (expansión «no saludable», dadas las características adipocitarias como elementos insulinorresistentes; ver panel inferior).")
Diferentes maneras por las que puede ocurrir la expansión de la masa de tejido adiposo, según una activación de la diferenciación normal de células progenitoras: hiperplasia (expansión «saludable», dadas las características adipocitarias como elementos respondientes a la acción de la insulina; ver panel superior) o a expensas del aumento del tamaño de los adipocitos maduros residentes en ese tejido: hipertrofia (expansión «no saludable», dadas las características adipocitarias como elementos insulinorresistentes; ver panel inferior).
. Esta disfunción tisular promueve múltiples disfunciones endocrino-metabólicas, como la insulinorresistencia, el estrés oxidativo a nivel del retículo endoplasmático, aumento de la actividad lipolítica, la hipoxia celular y la apoptosis. Estas alteraciones afectan a múltiples órganos: el hígado, el músculo, el páncreas endocrino y la función del endotelio. AGL: ácido graso libre; CRP: proteína C reactiva; DMT2: diabetes mellitus de tipo 2; EHGNA: enfermedad de hígado graso no alcohólica; IR: insulinorresistencia; JUNK: Janus kinase; NF-κB: NF kappa B; PHG: producción de glucosa hepática; RE: retículo endoplasmático; SNA: esteatohepatitis no alcohólica. Fuente: Adaptado de: Cusi K. Current Diabetes Reports. 2010;10:306.")
El tejido adiposo blanco e inflamación: consecuencias endocrino-metabólicas. La combinación de antecedentes genéticos, dieta poco saludable y estilo de vida sedentario puede inducir un aumento hipertrófico de la masa de tejido adiposo blanco y su infiltración por macrófagos, lo que conduce a un patrón anormal de producción (síntesis y liberación de adipocinas). Esta disfunción tisular promueve múltiples disfunciones endocrino-metabólicas, como la insulinorresistencia, el estrés oxidativo a nivel del retículo endoplasmático, aumento de la actividad lipolítica, la hipoxia celular y la apoptosis. Estas alteraciones afectan a múltiples órganos: el hígado, el músculo, el páncreas endocrino y la función del endotelio.
AGL: ácido graso libre; CRP: proteína C reactiva; DMT2: diabetes mellitus de tipo 2; EHGNA: enfermedad de hígado graso no alcohólica; IR: insulinorresistencia; JUNK: Janus kinase; NF-κB: NF kappa B; PHG: producción de glucosa hepática; RE: retículo endoplasmático; SNA: esteatohepatitis no alcohólica.
Fuente: Adaptado de: Cusi K. Current Diabetes Reports. 2010;10:306.
Tampoco se conoce con precisión cómo se hace la «distribución» de la energía y qué conduce al «reparto» de los lípidos para su asiento en tejidos adiposos de distintas partes del cuerpo, con tantas particularidades y tan diferente repercusión funcional, de tal manera que clínicamente aparece que la ubicación omental o perivisceral es la de mayor impacto metabólico (obesidad «central»). La experiencia con tiazolidinedionas muestra como estos agonistas de los PPAR (receptores nucleares que median diferentes efectos metabólicos por control de expresiones génicas), que se usan como agentes sensibilizadores de insulina (rosiglitazona y pioglitazona), promueven preferentemente la formación de tejido adiposo subcutáneo (con menor actividad metabólica) en vez de omental y disminuyen la grasa intramiocelular y hepática, que se refleja en la clínica, cuando con frecuencia decrecen las manifestaciones de insulinorresistencia mientras el paciente aumenta de peso93.
Alteraciones en el proceso oxidativoLa capacidad oxidativa influye sobre la cantidad de grasas en la célula e indica que la llegada de lípidos a los tejidos no es el único factor que influye sobre la resistencia de un órgano a la insulina. La plétora de ácidos grasos de cadena larga que no se logran oxidar también saturaría la capacidad del tejido adiposo para guardar y provocaría el escape de grasas desde los adipocitos hacia tejidos magros. Un mecanismo similar también afecta al músculo, que no puede desembarazarse de los lípidos que recibe.
Puede suceder que la sobrecarga de lípidos celulares exceda a la oxidación o que la maquinaria oxidativa no se active en tiempo o de la manera adecuada para quemar las grasas que llegan a la célula94. Aunque no se ha dilucidado cuál es la secuencia: se produciría un círculo vicioso en el que la disfunción mitocondrial, la elevación de los lípidos intramiocelulares, la alteración de la oxidación lipídica y la insulinorresistencia se perpetúen o se amplifiquen una con otra. Parece que la cadena de eventos podría comenzar con la alteración mitocondrial genética o con el aumento de los lípidos intramiocelulares en estados de insulinorresistencia desencadenada por factores ambientales.
Se demostró que la depresión de la oxidación grasa en roedores (por inhibición de la palmitoiltransferasa) aumentó la cantidad de lípidos celulares y promovió el estado de insulinorresistencia in vivo, con fuerte relación entre ambos fenómenos (r = 0,96)95.
Perseghin examinó la asociación entre sensibilidad a la insulina, oxidación lipídica y contenido intramiocelular de grasas en sujetos no obesos y aquellos que, dentro de su normopeso, tuvieron más contenido de grasa corporal y mostraron mayor capacidad oxidativa. Todos exhibieron sensibilidad a la insulina y contenido muscular de grasas normales. Esto indicó que el aumento de la oxidación de las grasas permite mantener la composición y cantidad celular de grasas y la respuesta a la insulina y revela que, en realidad, la homeostasis de la energía es un proceso integrado entre el ingreso (a través de nutrientes) y la oxidación de los productos metabólicos. Además que, entre los individuos, existe una amplia variación de la capacidad para oxidar el exceso de grasas dietarias.
Parte del fracaso de las dietas fuertemente restrictivas para el tratamiento de la obesidad se atribuye a que, en la medida en que se acentúa el menor ingreso, también desciende la oxidación de las grasas. Para mostrar la situación inversa, hay experimentos en los que se frustra el intento para inducir obesidad por la dieta, en modelos de ratas en las que se restringe la deposición de grasas (knock out de diacilglierol-aciltransferasa) u otro tipo de roedor en el que se promueve el aumento de la oxidación de grasas (knock out de acetil-CoA carboxilasa 2 o knock out de enzimas desacoplantes mitocondriales)96–98.
El ejercicio aumenta la capacidad oxidativa. Sin embargo, si se excluyen sujetos entrenados, el efecto de la actividad física sobre el contenido intramiocelular de grasas es motivo de controversia, ya que se ha observado que, si bien mejora la sensibilidad a la insulina y la oxidación de grasas, no modifica en forma significativa el contenido de lípidos intramiofibrilares. Esto indica que, al menos al principio, la mejoría de la insulinorresistencia por el ejercicio se produce por mecanismos que no incluyen la ectopia de la grasa muscular99,100.
Son diversas y complejas las explicaciones sobre los posibles fallos en las señales y la maquinaria de la oxidación, incluso con cambios que se han descripto en la biogénesis y función oxidativa y fosforilativa de las mitocondrias, en la insulinorresistencia y la DMT2. El grupo de Kelley midió mitocondrias mediante microscopia electrónica y observó que en el músculo esquelético eran más pequeñas en los diabéticos de tipo 2 y en los obesos y consideró que, además, tenían alteraciones en su capacidad bioenergética101.
En trabajos recientes, Wang et al. demostraron que, al inhibir la respiración y la fosforilación oxidativa por depresores de respiración o genes modificados incluidos en la biogénesis de las mitocondrias, puede alterarse la diferenciación de preadipocitos y la respuesta de los adipocitos a la insulina. Más aún, el defecto mitocondrial podría causar una disminución en la secreción de adiponectina que provoque una declinación en la utilización de la glucosa de otros tejidos102.
La disfunción mitocondrial disminuye la posibilidad de eliminar AG dentro de los adipocitos por desacoplamiento de la β-oxidación y por reesterificación, mientras que la actividad de la propia mitocondria promueve sustratos para la producción de glicerol103.
Se ha asociado la disminución de la actividad fosforilativa oxidativa mitocondrial al aumento de lípidos intramiocelulares y se ha revisado desde la actividad de las enzimas oxidativas hasta la cadena de transporte de electrones en personas con DMT2. Se considera que hay evidencias de la disminución de la actividad de enzimas oxidativas en el músculo esquelético de estos pacientes. El fallo es independiente del tipo de fibra muscular y se acompaña de daño mitocondrial y, en la mayor parte de los ensayos, la menor actividad oxidativa de las enzimas se explica por el descenso en el contenido de mitocondrias y aún no convence la hipótesis de un trastorno funcional de las mitocondrias en los diabéticos.
Aunque las modificaciones morfológicas y en la función oxidativa pueden reflejar una vida sedentaria que puede cambiar por la actividad física, los estudios sobre insulinorresistencia en familiares directos de diabéticos de tipo 2 han aportado evidencias sobre los defectos metabólicos tempranos en la DMT2, entre los que se incluyen una baja producción basal de ATP y una pobre respuesta al estímulo con insulina. Hay quienes no se adhieren en su totalidad a los conceptos anteriores, pues estiman que no hay pruebas de que la producción basal disminuida afecta la totalidad de la oxidación de lípidos y glúcidos, de allí que aún no existan suficientes evidencias sobre las causas y los mecanismos que reúnen al trastorno oxidativo mitocondrial y la insulinorresistencia y, menos aún, el efecto específico del entrenamiento físico y el trabajo muscular sobre la función y la plasticidad mitocondrial en la DMT2104.
Siempre existe la especulación de un vínculo patogenético entre la sobrealimentación y el sedentarismo, o sea, de la homeostasis del ingreso y el gasto energético y, en ese sentido, la función central de la mitocondria en la utilización y la producción de energía la ubican en una incómoda posición para explicar el impacto que puede tener su disfunción sobre el equilibrio metabólico del organismo. Así, la hipótesis de una mitocondria insuficiente o disfuncional es seductora, aunque aún incierta. Se piensa que es posible que haya alteraciones genéticamente determinadas o alteraciones vinculadas a la inactividad que modifiquen la actividad oxidativa de respuesta de adaptación a la sobrealimentación, con cúmulo crónico de metabolitos de la oxidación que pueden favorecer la resistencia a la insulina o la disfunción celular105.
Se acepta en forma general que el estrés oxidativo contribuye al daño celular y tisular glucolipotóxico de la diabetes. El origen de las especies de oxígeno reducido en las células β del páncreas y en tejidos insulinosensibles es la cadena de transporte de electrones de las mitocondrias106.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónEl estudio fue financiado por el Proyecto FPREDM-052015 (Fondation pour la Recherche en Endocrinologie, Diabetologie et Metabolisme, Lausanne, Suiza).
Conflicto de interesesLos autores declaran que no existe conflicto de interés alguno y que ellos son los responsables del contenido y de la escritura del manuscrito.