








Existe una serie de síndromes caracterizados por poliposis intestinal, que tienen marcadores cutáneos característicos. Con sentido didáctico y pronóstico, podemos dividirlos según indica la tabla I.
Esta división es importante, pues diferencia los pólipos verdaderos de los seudopólipos (como en las neurofibromatosis, que si bien en el estudio de imágenes parecen pólipos, se trata de neurofibromas que asientan en la mucosa); y los adenomatosos (con alto potencial de malignización) de los hamartomatosos (con extremadamente bajo potencial degenerativo).
En la actualidad se les agrupa según el gen causal y su expresión fenotípica (tabla II).
SÍNDROME DE GARDNEREste síndrome fue descrito en 1912, pero es en 1953 que Gardner y Richards1 le dan ubicación nosológica. Se caracteriza por la presencia de pólipos intestinales, quistes epidérmicos y osteomas. Es una variante fenotípica de la poliposis adenomatosa familiar (PAF); su incidencia es de 1:8.000 a 1:16.000 nacidos vivos2 .
Se transmite en forma autosómica dominante y exhibe alta penetrancia. El gen causal es el APC (adenomatous polypsosis coli) que codifica una proteína de 2.843 aminoácidos y mapea en el cromosoma 5q21. Dicho gen es la causa de diferentes expresiones fenotípicas: poliposis adenomatosa familiar, síndrome de Gardner y síndrome de Turcot (asocia poliposis colónica y tumores de sistema nervioso central)2-4 .
El gen APC actúa como supresor tumoral. Uno de los dominios mejor caracterizados del gen APC interacciona con la β catenina, lo que permite una función en la adhesión y el desarrollo celular. Se ha propuesto que, al estar mutado, el gen APC favorecería el crecimiento tumoral, por desregulación de la cascada de la β catenina 5,6 . Existe una correlación genotipo-fenotipo; de acuerdo con el lugar donde se produce la mutación varía la expresión clínica de la enfermedad. Si la mutación se localiza en el tercio inicial o final del gen se asocia a una poliposis moderada, con manifestaciones tardías; si, en cambio, la mutación afecta a su parte central desarrolla enfermedad severa, con gran número de pólipos, asociada a manifestaciones extracolónicas a temprana edad 2,5,6 .
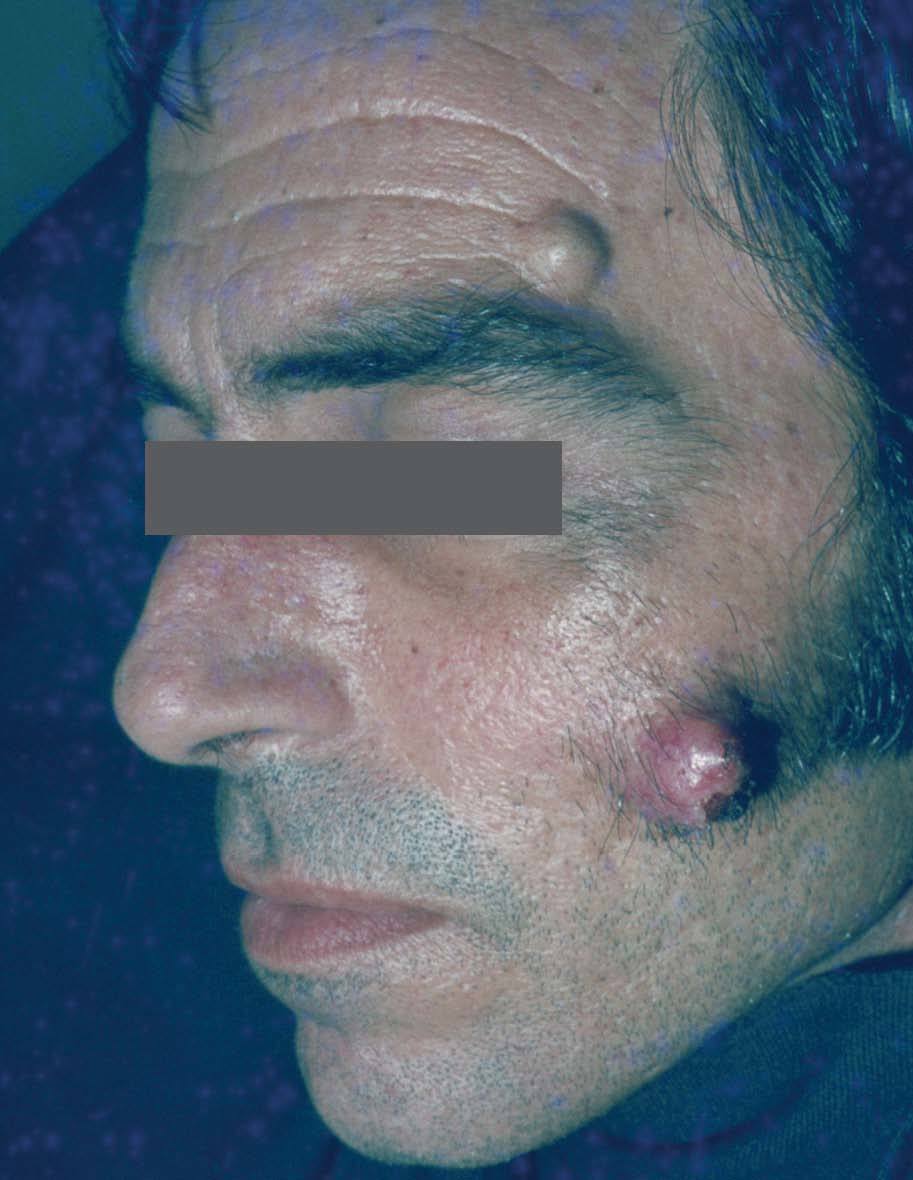
Manifestaciones cutáneasLos quistes epidérmicos son la principal manifestación cutánea. Se observan en el 50 al 100% de los casos. En general aparecen en la infancia, son múltiples y se localizan en piernas, cara y cuero cabelludo (a diferencia de los quistes epidérmicos no sindrómicos que se localizan en dorso)7-10 (fig. 1).
Su histología es similar a la de casos aislados y se caracteriza por una lesión quística, localizada en la dermis, compuesta por una pared de epitelio escamoso estratificado con presencia de granulosa, y una cavidad con contenido de queratina. En dos tercios de los pacientes se puede observar un aspecto similar a pilomatrixoma11 . Estos quistes tienen un curso benigno, y requieren exéresis sólo cuando presentan un problema estético, o cuando se rompen y provocan inflamación secundaria.
Los fibromas pueden aparecer en piel, tejido subcutáneo, mesenterio o retroperitoneo. Los tumores desmoides son característicos y se desarrollan en un 9% de los pacientes, se localizan en piel y tejidos blandos de pared abdominal anterior y en retroperitoneo. Se presentan en pacientes jóvenes con una edad promedio de 29,8 años; afectan con mayor frecuencia a las mujeres, con una relación mujer:varón de 3:1 7-9 . La histología evidencia un tumor poco circunscrito, compuesto en su mayoría por células fusiformes, con atipia ausente o mínima. Si bien no metastatizan, tienen un comportamiento agresivo e invasivo local. Tienden a recurrir luego de la extirpación quirúrgica.

TABLA I. Poliposis intestinal con marcadores cutáneos

TABLA II. Genes implicados en los cuadros de poliposis intestinal hereditaria
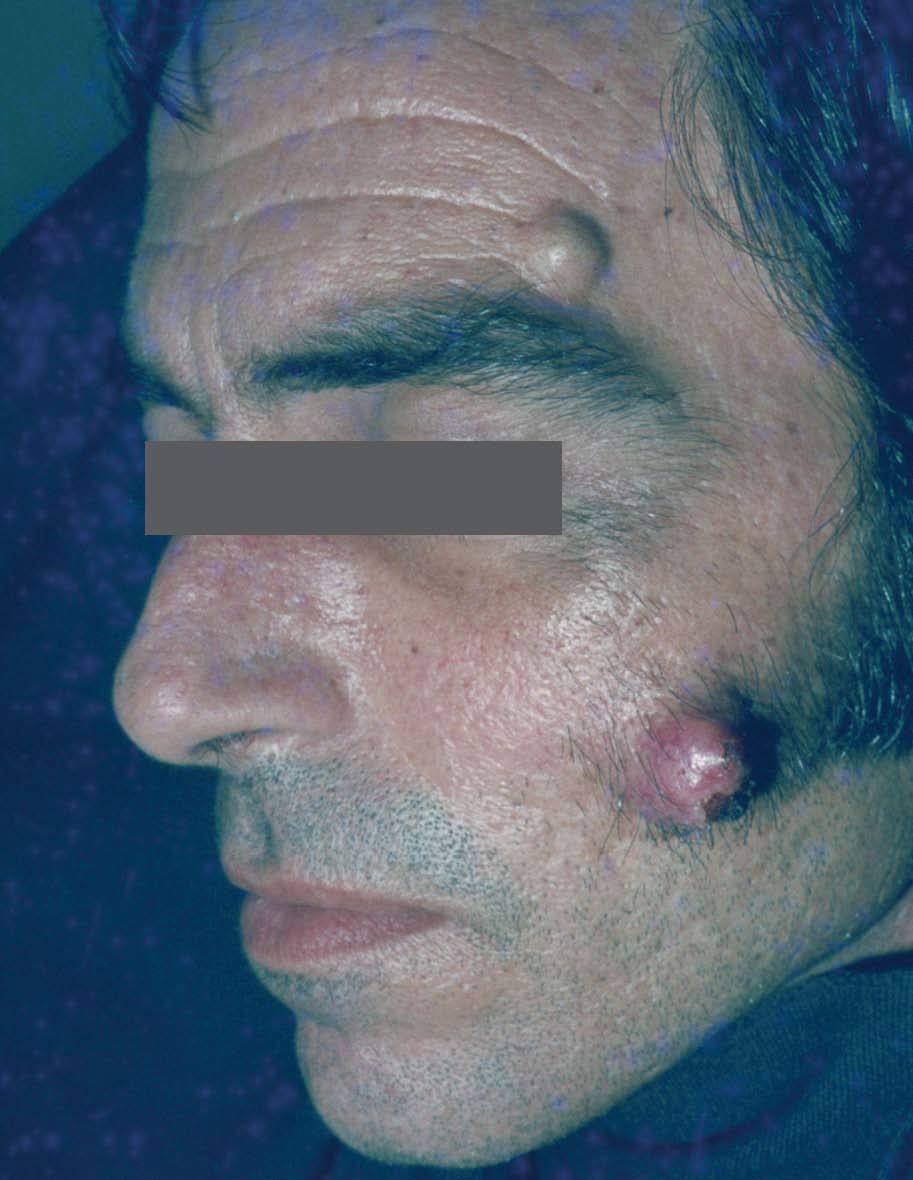
Figura 1. Quistes en la cara. Síndrome de Gardner.
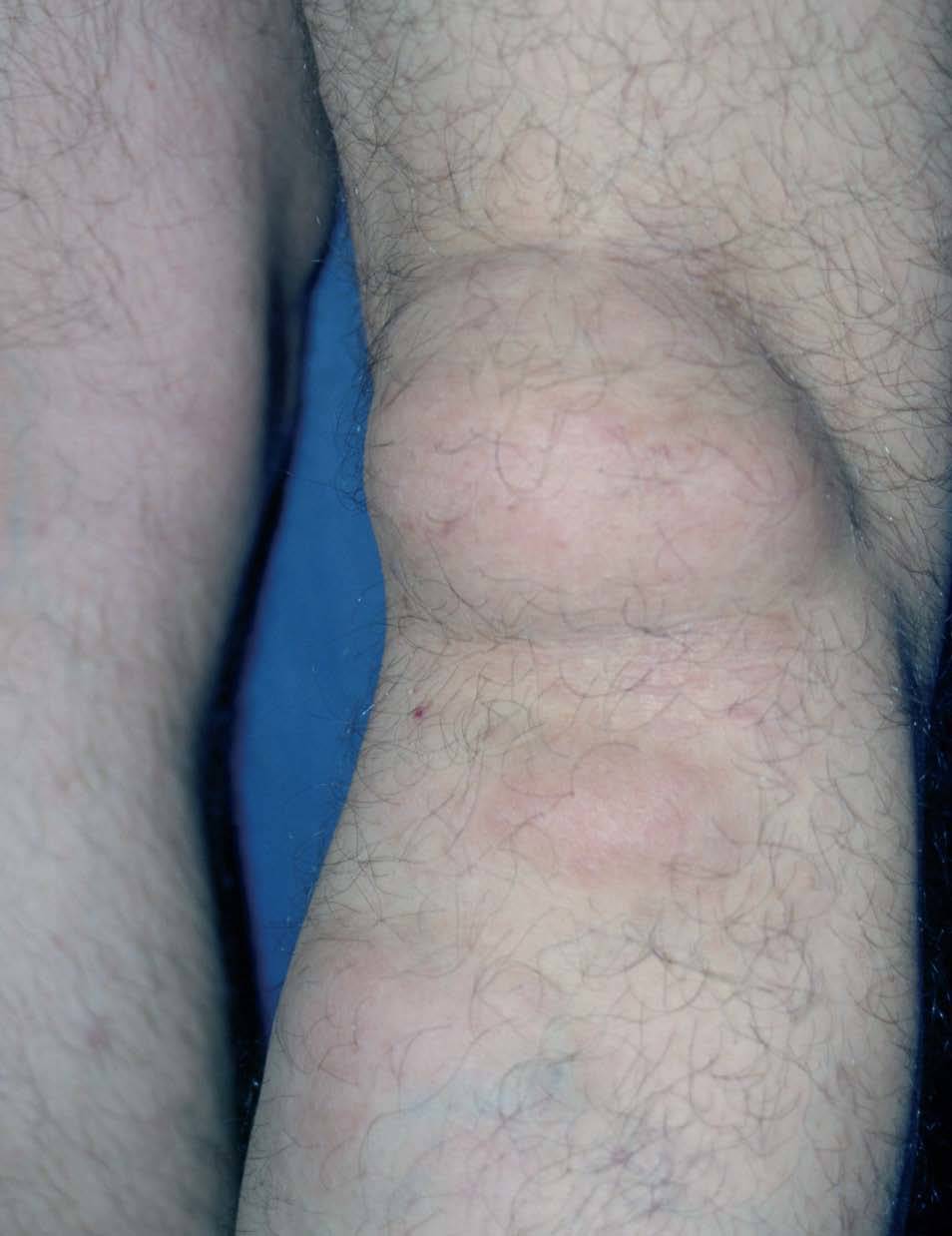
Otras alteraciones cutáneas observadas incluyen: lipomas (fig. 2), leiomiomas, neurofibromas y lesiones pigmentadas de piel 8-10 .
Manifestaciones extracutáneasLas alteraciones oculares se presentan en el 90% de los pacientes, y son el primer hallazgo diagnóstico. Se caracterizan por la presencia de hipertrofias congénitas del epitelio pigmentado retiniano (CHRPE). Se presentan en forma bilateral, en mayor número y con pigmentación mixta (comparado con formas esporádicas) y se las considera un marcador específico de PAF. Es por este motivo que se recomienda un examen oftalmológico temprano12,13 .
Los osteomas son un hallazgo esencial del síndrome, y se observan al menos en el 50 al 75% de los casos; con incidencia en los huesos de la cara, en especial la mandíbula y el maxilar. Aparecen en la infancia, y preceden a la poliposis intestinal. Alcanzan su tamaño definitivo en algunos años, y luego continúan provocando cambios graduales aun en el adulto. Son neoplasias benignas, bien diferenciadas. Pueden dividirse en dos tipos: centrales, que se desarrollan a partir del endostio, y periféricos, cuando se desarrollan del periostio. Para su detección se recomienda realizar una radiografía panorámica periódica de mandíbula, si bien la tomografía computa-rizada (TC) es un método más sensible de cribado. La escoliosis ha sido considerada parte del síndrome. También puede haber exostosis, endostosis y espesamiento cortical de los huesos 9,10,14,15 .
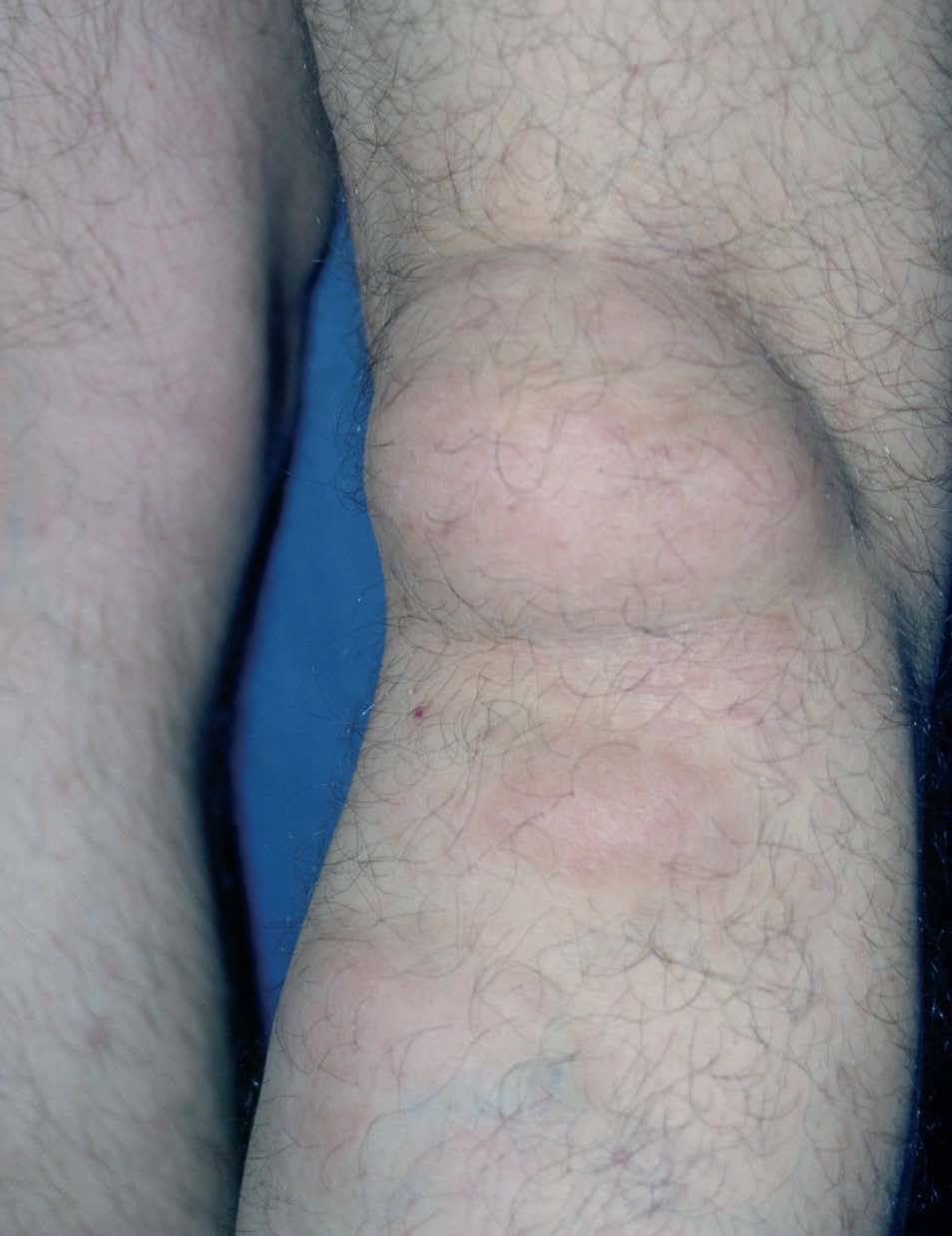
Figura 2. Lipoma de gran tamaño en hueco poplíteo. Síndrome de Gardner.
Las anomalías dentales están presentes en un 18% de los casos. Son un signo temprano y se manifiestan con un espectro variable de alteraciones que incluyen: ausencia congénita de dientes, dientes supernumerarios e impactados en ambos maxilares, fusión de raíces dentales, multicaries, odontomas y quistes 6,9,10 .
La poliposis intestinal es el rasgo más importante del síndrome; se presenta en el 100% de los pacientes. Se desarrolla en la pubertad, con una edad media de 22 años, y va aumentando en número con el tiempo. Se localiza en el colon y el recto, con frecuencia en los pliegues sigmoideo, esplénico y hepático. Pueden observarse con menor frecuencia en el intestino delgado y el estómago. Estos pólipos tienen un 100% de posibilidad de desarrollar adenocarcinoma; usualmente lo hacen en la tercera década de la vida7,9,10,15 . Es por este motivo que se recomienda realizar estudios periódicos de sangre oculta en materia fecal, sigmoideoscopia, colonoscopia y endoscopia del tracto digestivo superior. La colecto-mía se recomienda ante la presencia de 30 o más pólipos, o cuando hay displasia o malignidad en un pólipo.
Si se preserva el recto, existe del 25 al 59% de posibilidad de desarrollar un carcinoma rectal tras la colecto-mía, motivo por el cual se recomienda también la resección completa de la mucosa rectal9,10 .
El segundo cáncer más común es el carcinoma periampular, que se observa en el 12% de los casos, desarrollado a partir de pólipos duodenales cercanos a la ampolla de Vater2,7,9 .
Se han descrito otros tumores asociados de páncreas, vejiga, tracto biliar, etc.
El cáncer de tiroides tiene mayor frecuencia con respecto a la población general y se produce a edades más tempranas. Generalmente son carcinomas papilares, que se desarrollan a una edad promedio de 23,6 años, más frecuente en mujeres. Por este motivo se recomienda un examen tiroideo y ultrasonografía periódica, y biopsia en lesiones mayores de 1 cm de diámetro9 .
SÍNDROME DE MUIR TORREDe forma independiente, Muir et al16 , en 1967, y Torre17 , en 1968, describieron los primeros casos del síndrome que desde entonces lleva sus nombres. Se define por la presencia de, al menos, un tumor de glándulas sebáceas y una neoplasia interna (con o sin queratoacantoma)18 .
El diagnóstico se realiza a una edad promedio de 54 años, es más frecuente en el varón (2:1) con respecto a la mujer19,20 .
Se trata de una genodermatosis que se transmite en forma autosómica dominante con penetrancia incompleta y expresión clínica variable. En la actualidad se le considera una variante de HNPCC (cáncer colorrectal no polipósico hereditario), en la cual existe afección cu-tánea21,22 . Está causado por la mutación germinal de al menos cuatro genes que codifican proteínas relacionadas con la reparación del ADN (MMR): MSH2 (cromosoma 2p16), MLH1 (cromosoma 3p21), PMS2 (cromosoma 7p22), MSH622,23 .
En el síndrome de Muir Torre se detectan mutaciones en un 40% de los casos, las cuales en su mayoría se localizan en el gen MSH2 . Estas mutaciones determinan una falla en la reparación del ADN, y produce inestabilidad genética, que se puede evidenciar mediante la prueba de inestabilidad de microsatélites24 .
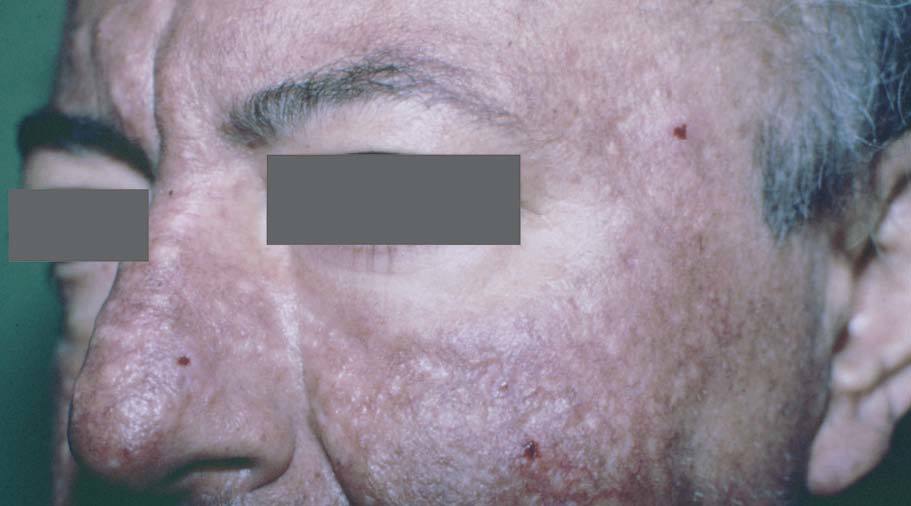
Manifestaciones cutáneasLos tumores sebáceos son el signo cutáneo principal y algunos autores los consideran un pilar diagnóstico. Pueden preceder a la malignidad interna en un 22%, aparecer en forma concomitante en un 6% y con posterioridad en un 54%19,20 (fig. 3).
Se manifiestan con un espectro clínico que incluye: hamartoma sebáceo, adenoma sebáceo, epitelioma sebáceo y carcinoma sebáceo. Algunos autores incluyen también al carcinoma basocelular con diferenciación sebácea. De ellos el más frecuente y específico del síndrome es el adenoma sebáceo que se caracteriza clínica-mente por la presencia de múltiples pápulas o nódulos amarillentos de consistencia firme, frecuentemente localizados en la cabeza y el cuello. Se desarrollan en edad adulta, si bien hay descrito casos infantiles 7,10,20 . Desde el punto de vista histológico se reportan como lesiones de difícil clasificación entre las entidades clásicas; si bien la presencia de adenomas sebáceos con signos de queratoacantomas o con imágenes quísticas debe hacer sospechar el diagnóstico de este síndrome 25 .
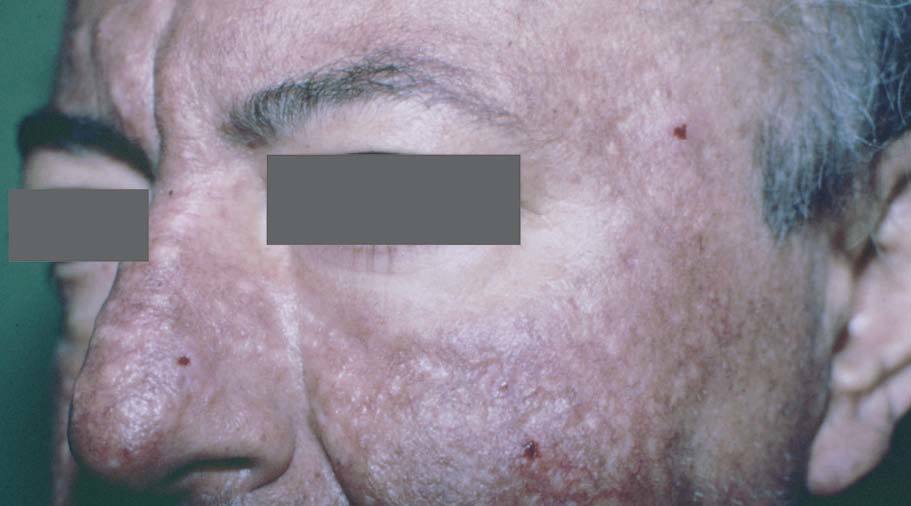
Figura 3. Hiperplasia sebácea y adenoma sebáceo. Síndrome de Muir Torre.
El carcinoma de glándulas sebáceas es otro hallazgo frecuente. Puede ser de origen extraocular o desarrollarse a partir de las glándulas de Meibonio. Éste, a diferencia del extraocular, tiene mayor frecuencia de metástasis. Histológicamente muestran gran variabilidad 20 .
En un 30 a un 50% de los casos se observa la presencia de queratoacantomas múltiples, que se localizan en la cabeza y el cuello. Aun en ausencia de adenomas sebáceos, con la presencia de queratoacantomas asociados a neoplasia visceral algunos autores sugieren el diagnóstico de Muir Torre25 .
Entre los métodos diagnósticos se propone el estudio de inestabilidad de microsatélites, presente en el 86% de los tumores de estos pacientes, a diferencia del 15% en tumores esporádicos. Además, ante la presencia de lesiones de origen sebáceo con hallazgos inusuales, se propone el análisis inmunohistoquímico de la pieza para marcar la expresión de las proteínas hMSH2 y hMLH125 . Este método puede identificar en alto porcentaje los portadores de mutaciones germinales en genes MMR y puede ser utilizado para cribado en familiares, pero no reemplaza al test de inestabilidad de microsatélites26 .
Manifestaciones extracutáneasEl carcinoma colorrectal se desarrolla en un 61% de los casos, y se localiza con mayor frecuencia en el ángulo esplénico. Se caracteriza por ser hereditario, y se desarrolla en el contexto de pólipos colónicos benignos y escasos en número. Se presenta en un promedio de edad menor que en la población general, y tiene un curso indolente en la mayoría de los casos, dada su larga sobrevida después del diagnóstico21,22 .
En orden de frecuencia se observa cáncer del tracto urinario (22%), cáncer de mama (6%), malignidad hematológica (5%) y carcinoma de intestino delgado (3,5%); se han descrito también tumores de laringe, ampolla de Vater, riñón, útero y ovario19,20 .
Se proponen para el seguimiento de los pacientes: colonoscopias cada 2 años a partir de los 25 años, y anual para los portadores conocidos; estudio ginecológico (con punción-aspiración endometrial, ultrasonido, PAP y examen mamario y pélvico) cada año después de los 30 años de edad. Para la detección de cáncer del tracto urinario: ultrasonido, citoscopia y análisis de orina cada año 21 .
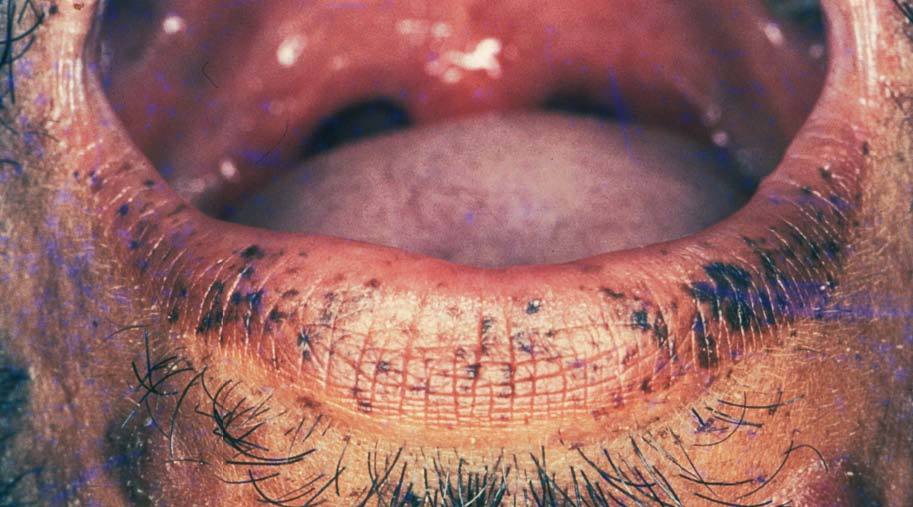
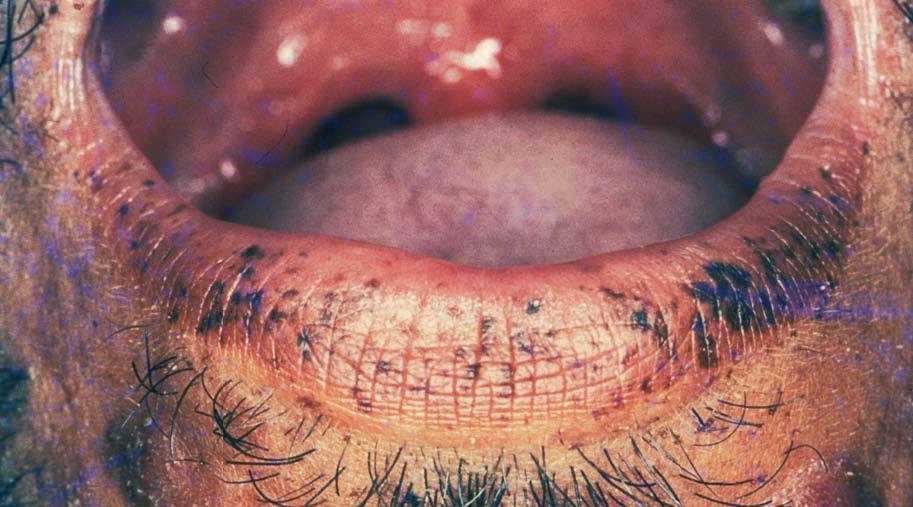
Figura 4. Máculas pigmentadas en el labio inferior. Síndrome de Peutz-Jeghers.
SÍNDROME DE PEUTZ-JEGHERS
Este síndrome fue descrito por Peutz, en 1921, como un síndrome hereditario que asociaba poliposis intestinal y pigmentaciones mucosas. Jeghers et al, en 1949, presentaron dos casos en la bibliografía inglesa, desde entonces se conoce esta entidad como síndrome de Peutz-Jeghers (SPJ)7,27 . Su incidencia se estima en 1:8.300 a 1:29.000 nacidos vivos28 .
Se hereda en forma autosómica dominante, y se han descrito mutaciones frescas28 . El gen involucrado en el SPJ se localiza en el cromosoma 19p13.3, y se denomina STK1/LKB1; está compuesto por 10 exones, y codifica una proteína treoninacinasa de 433 aminoácidos. Este gen interviene en el ciclo celular G1; en la vía de apoptosis p53 dependiente; en la regulación del factor de crecimiento del endotelio vascular, y en la vía Wnt-β catenina. La mayoría de las mutaciones de este gen (deleciones, inserciones y reordenamientos) dan como resultado una proteína truncada, sin actividad de cinasa29-31 . Por lo tanto el SPJ representa el primer síndrome con susceptibilidad al cáncer, resultante de la mutación germinal que inactiva la actividad de la cinasa. Las mutaciones germinales se detectaron sólo en un 50-70% de los casos familiares y en un 12-50% de los casos esporádicos. Existen diferentes hipótesis que intentan explicar por qué no se halla la mutación en todos los individuos afectados; una de ellas propone la falta de detección de la mutación debido a la existencia de alteraciones genéticas no detectables por lo métodos de estudios habituales. Otros autores proponen un segundo locus para el SPJ localizado también en el cromosoma 19p, donde mapea el gen STK1329,30 .
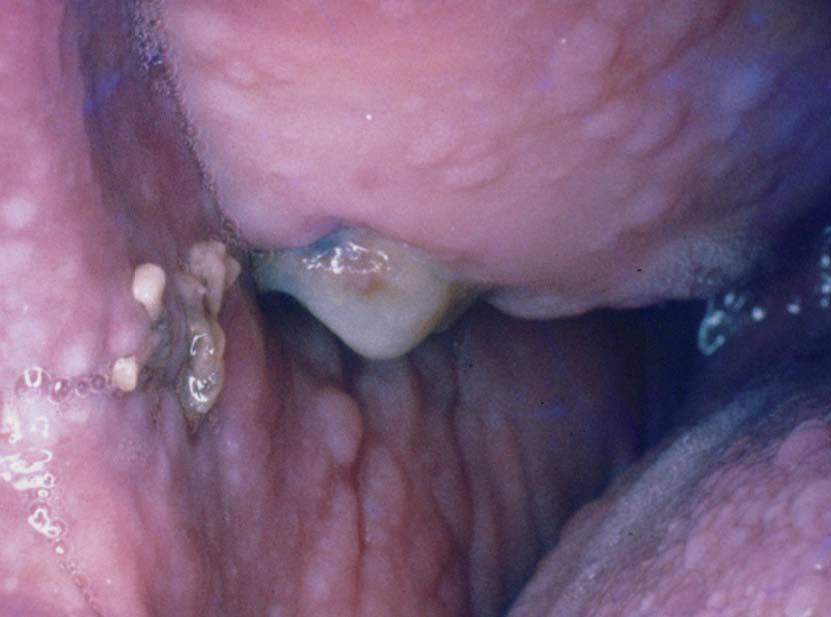
Manifestaciones cutáneasLas pigmentaciones mucosas son la clave diagnóstica. Se caracterizan por la presencia de máculas que varían entre 2 a 12 mm de diámetro, de color marrón oscuro, marrón azuladas o azules dependiendo del tipo racial27,28,32 . Se localizan preferentemente en los labios (96%) con predominio del inferior y en la mucosa oral (83%) (fig. 4). Las encías y el paladar pueden estar afectados, mientras que la lengua y las fauces, en general, están libres de lesión. Los párpados suelen estar afectados (36%), y en menor frecuencia puede tomar la conjuntiva. En la cara, las lesiones tienden a confluir alrededor de la boca y la nariz 7,32,33 .
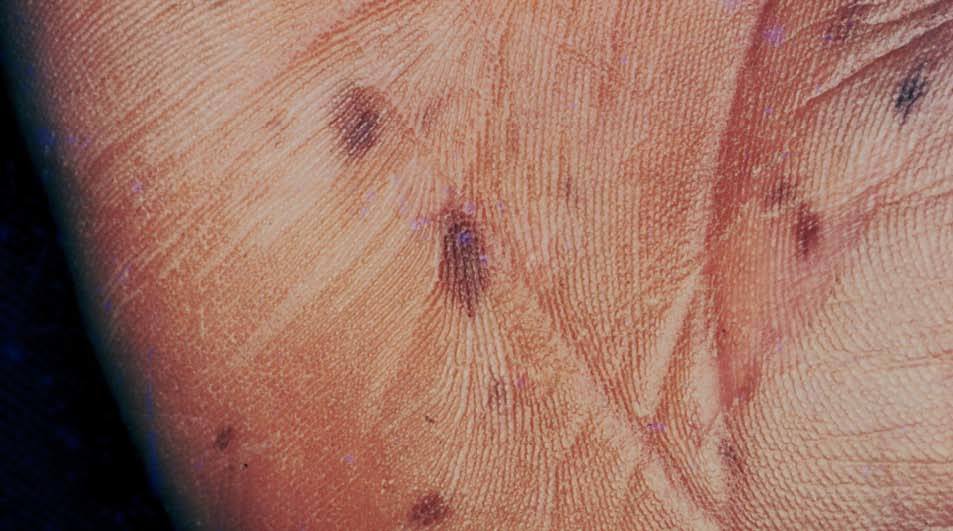
Figura 5. Máculas pigmentadas en la palma. Síndrome de Peutz-Jeg-hers.
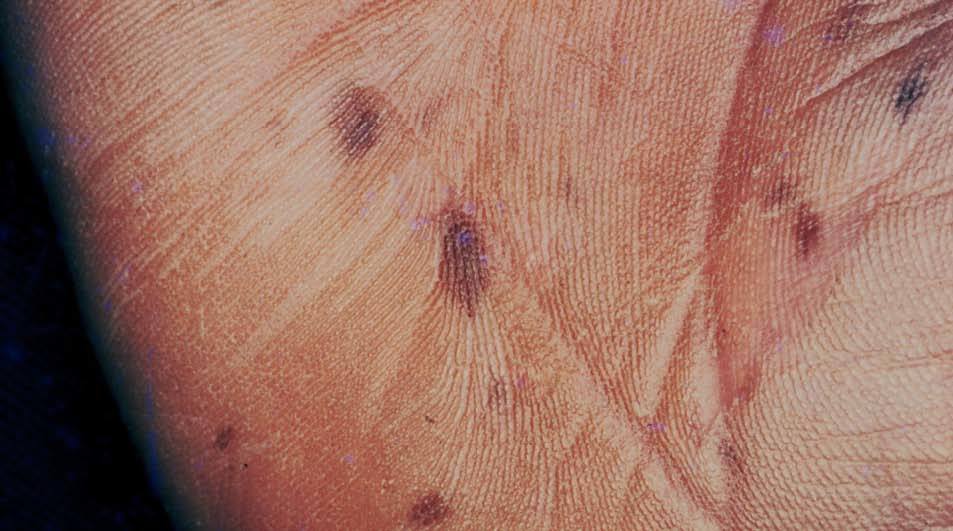
Las palmas y plantas están comprometidas en un 32% de los casos, y pueden resultar afectados los dedos de manos y pies (sin borrar los dermatoglifos) (fig. 5)32 . Otras localizaciones afectadas son: región periumbilical y perianal, dorso de manos.
Las máculas se desarrollan a partir de los 2 años de edad (si bien existen casos congénitos y otros desarrollados en la adultez), y alcanzan su máxima expresión en la pubertad. En general, las máculas de localización cutánea y labial tienden a desaparecer con los años, mientras que las de localización mucosa persisten toda la vida32 .
Existe una amplia expresión fenotípica de las máculas entre las familias afectadas. Pueden variar desde máculas solitarias hasta múltiples, con diferentes tonalidades del marrón. Esto puede atribuirse a diferentes expresiones del gen afectado. No se ha observado correlación entre la localización y la intensidad de las máculas con respecto a la gravedad de la poliposis. Reportes iniciales mencionan disociación de síntomas, por lo cual individuos afectados en diferentes generaciones en una misma familia pueden presentar poliposis y pigmentaciones o ambas 28,32 .
Estas asociaciones dependen de la edad; en la infancia sólo pueden observarse pigmentaciones; en la adultez temprana, pigmentaciones con pocos síntomas intestinales y, en el adulto, gran deterioro intestinal con afección cutánea escasa. De allí la importancia del examen de mucosa oral para establecer el diagnóstico7 .
Desde el punto de vista histológico, las máculas se caracterizan por el aumento de cantidad de melanina en las células de la capa basal (pecosidades).
Estudios de microscopia electrónica de máculas labiales revelan melanosomas dispersos, muy pigmentados, mientras que en las pigmentaciones digitales se encuentra un bloqueo pigmentario, con muchos melanosomas en las dendritas de los melanocitos y muy pocos en los queratinocitos32 .
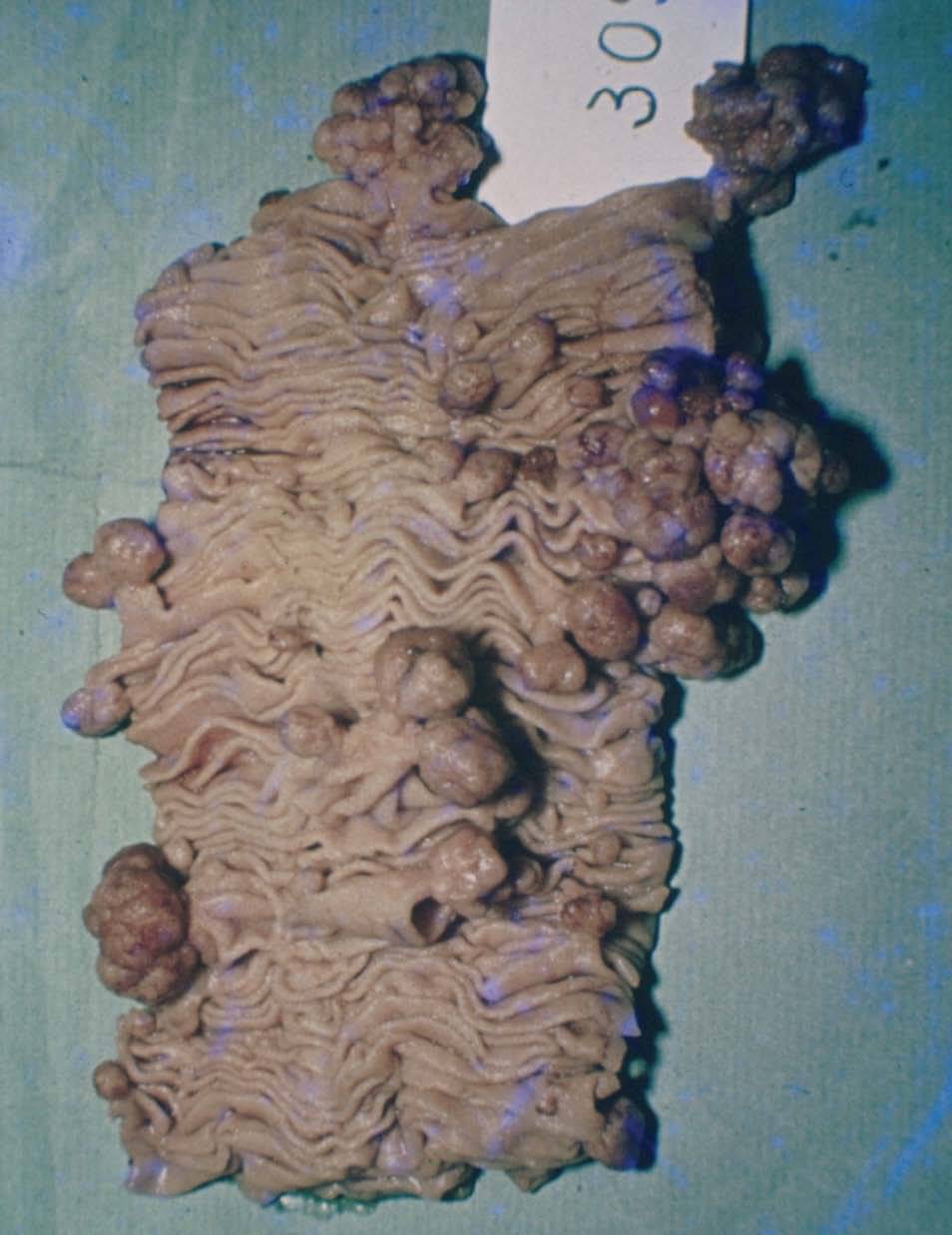
Manifestaciones extracutáneasLa clave diagnóstica del síndrome de Peutz-Jeghers es la presencia de pólipos hamartomatosos a lo largo del tracto digestivo, en particular en el intestino delgado (64-96%), principalmente en el yeyuno e íleo (fig. 6)7,28 . También se localizan en el colon (60%), el recto, el estómago (24-29%) y el duodeno e incluso en el apéndice; el esófago está libre. El número es variable; se han descrito pólipos solitarios, si bien en el 90% de los casos son múltiples, particularmente en personas mayores. El tamaño varía desde milímetros a 3 cm; los pequeños son sésiles, y los grandes, usualmente, pedunculados. Los pólipos pueden aumentar en número y tamaño con patrones de crecimiento rápido intercalados con períodos de quiescencia que pueden durar de meses a años. Los pólipos grandes causan intususcepción del intestino, especialmente en el delgado28 .
Desde el punto de vista histopatológico, los pólipos son hamartomas, determinados por el crecimiento aberrante de tejido normalmente hallado en ese sitio. Se caracterizan por la presencia de un eje arborescente de tejido conjuntivo y músculo liso revestido por epitelio intestinal normal, rico en células caliciformes. La presencia de las células de Paneth es un hallazgo diferencial entre los pólipos hamartomatosos de Peutz-Jeghers y los pólipos adenomatosos. Si bien se observa actividad mitótica en los pólipos, la displasia no es usual28,30 .
La seudoinvasión es otro hallazgo histológico que se puede observar; preferentemente en el intestino delgado, y debe diferenciarse del adenocarcinoma mucinoso.
Si bien los pólipos se presentan en mayor grado en el intestino, existen reportes de pólipos en otros órganos: nasal, uretral, vejiga, tracto biliar32 .
Aproximadamente el 33% de los pacientes presentan sintomatología en la primera década de la vida y un 60% entre los 15 y 18 años de edad. La consulta más frecuente es el dolor abdominal tipo cólico intermitente que se produce luego de las comidas. Los episodios de dolor pueden recurrir en intervalos de semanas o meses, se tornan más severos con la edad, con remisiones más cortas. Su intensidad depende del grado y localización de los pólipos, debido frecuentemente a intususcepción intestinal (86%)34 .
Una complicación que surge en la segunda o tercera década de la vida son ataques de intususcepción de intestino delgado con obstrucción. Este cuadro a menudo se resuelve espontáneamente y en ocasiones requiere cirugía.
Otras complicaciones observadas con frecuencia son el sangrado rectal, en el 81% de los pacientes, y la hematemesis, en un 10%, causante de la anemia ferropénica7 .
Una complicación habitual en edades pediátricas es la extrusión anal de los pólipos con prolapso rectal, aun antes de desarrollarse las pigmentaciones. En adultos, la extrusión anal de los pólipos se observa en un 24% de los casos.
Los pacientes con el síndrome de Peutz-Jeghers tienen un riesgo aumentado para desarrollar cáncer con respecto a la población general7 .
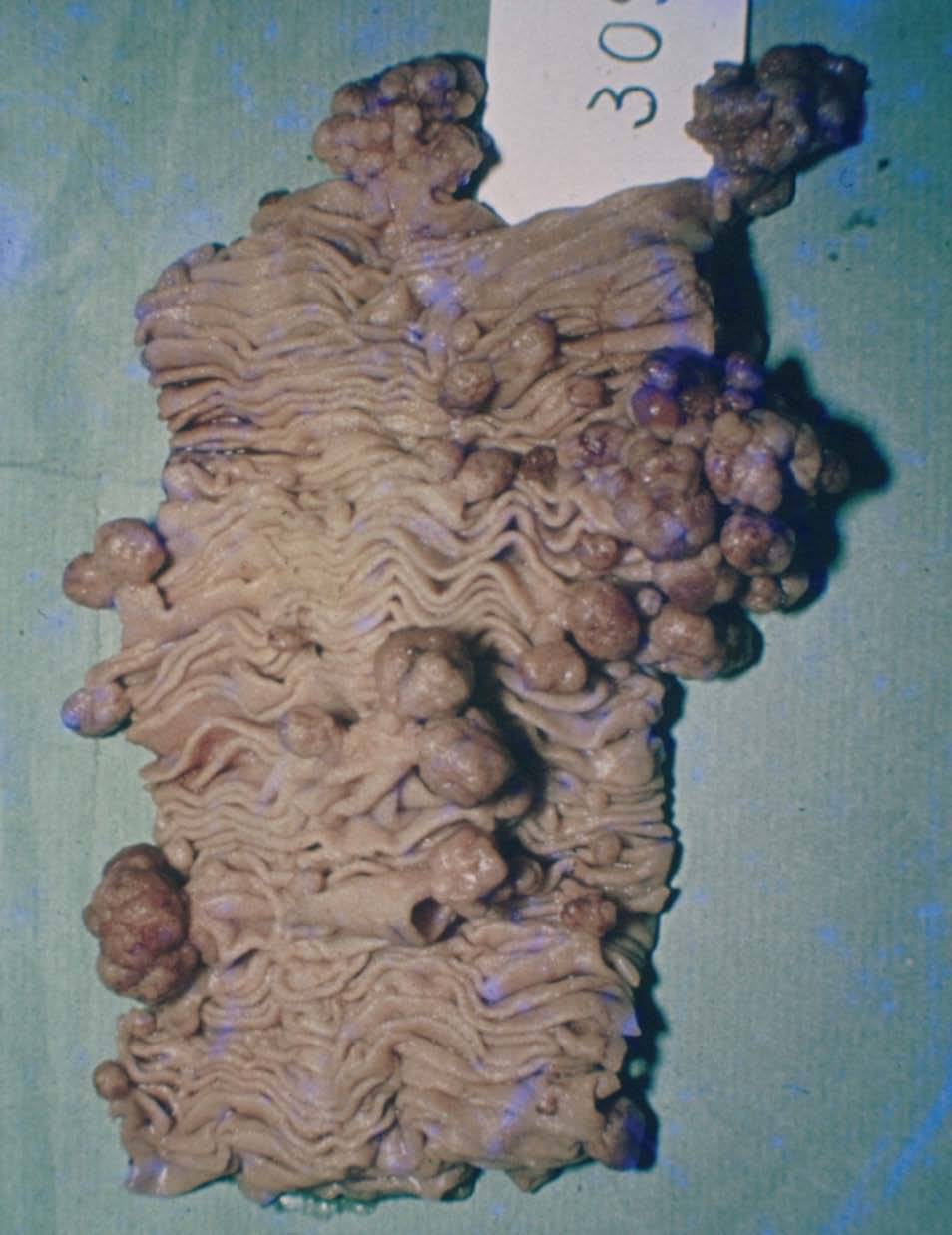
Figura 6. Macroscopia de pólipos en intestino delgado. Síndrome de Peutz-Jeghers.
Los pólipos hamartomatosos no tienen potencial maligno, si bien existen reportes de cáncer gastrointestinal en estos pacientes. Algunos autores consideran que pueden surgir de una mala interpretación de seudoinvaginación que simula un adenocarcinoma mucinoso. Otros refieren que el desarrollo de la neoplasia intestinal (estómago, colon) no está relacionada con la localización predominante de los pólipos (intestino delgado). Hay quienes argumentan la coexistencia de adenomas, causantes del desarrollo de la neoplasia ulterior. Finalmente, existen reportes de desarrollo de neoplasia sobre pólipos hamartomatosos, con cambios adenomatosos y progresión al adenocarcinoma7,28 .
La malignidad del tracto gastrointestinal se observa en un 2-3% de los casos; la más frecuente es la del intestino grueso, seguido por el estómago, en una edad media de 50 años. Estas neoplasias se localizan en iguales áreas que en la población general. Sin embargo, se han reportado adenocarcinomas de yeyuno e íleo en pacientes con SPJ (malignidad extremadamente poco frecuente en la población general)28,34 .
Las mujeres con SPJ pueden desarrollar en un 5 a un 12% de los casos diferentes tipos de tumores de ovario, entre los cuales cabe destacar el tumor del cordón sexual con túbulos anulares32 . Este tumor es raro en la población general y llamativamente frecuente en el SPJ y es por eso que algunos autores lo consideran una manifestación fenotípica del síndrome. Se pueden establecer diferencias en ambas poblaciones; en los casos aislados no sindrómicos el tumor es unilateral, de gran tamaño y no calcificado, con comportamiento maligno y tendencia a la metástasis. El asociado al SPJ es bilateral, de tamaño microscópico con distribución multifocal, y con depósito de calcio; el tumor tiene curso benigno y, dado que es un tumor funcionante, puede asociarse a precocidad sexual. En mujeres adultas produce menstruación irregular o sangrado posmenopáusico, y en ocasiones hiperplasia endometrial35,36 .
Otra neoplasia que puede desarrollarse es el cistoadenoma mucinoso con metaplasia del epitelio tubular.
El carcinoma mamario ha sido reportado frecuentemente en mujeres con SPJ; a menudo se presenta en forma bilateral y en la premenopausia (hecho poco frecuente en la población general). Desde el punto de vista histológico, en general son carcinomas infiltrantes intraductales, si bien se han reportado casos de carcinoma papilar. En estos pacientes, los carcinomas de mama estarían estimulados por la producción de estrógenos7,28 .
Se ha observado también adenoma maligno o carcinoma con desviación mínima de endocérvix, que es un adenocarcinoma bien diferenciado. Este tumor tiene un comportamiento maligno, con rápida progresión y diseminación temprana. Responde mal a los tratamientos convencionales y es de mal pronóstico. Es raro en la población general y relativamente frecuente en pacientes con SPJ, lo que sugiere una relación patogénica entre ambos36 .
Otro tumor raro en la población general, pero frecuente en el SPJ, es el tumor de las células de Sertolli. Se presenta en forma multifocal y bilateral en testículos. Se considera el equivalente en el varón del tumor del cordón sexual con túbulos anulares femenino37 .
Se debe realizar endoscopia alta y baja y radiografía seriada de intestino delgado, con un intervalo recomendado de 2 años; con remoción por esta vía de los pólipos sintomáticos o mayores de 1,5 cm. Los pólipos localizados en el intestino delgado son de difícil acceso por vía endoscópica; por este motivo, para su extirpación se realiza cirugía convencional28 .
Debe ser de rutina el control ginecológico y el examen mamario.
Las máculas labiales, cuando constituyen un problema estético importante, pueden ser removidas con láser.
Dada la alta susceptibilidad a cáncer de mama y ovario se requiere un examen ginecológico de rutina, además del autoexamen de mama28 .
SÍNDROME HAMARTOMATOSO-TUMORAL DEL PTENEl gen PTEN (también denominado MMAC1 o TEP1 ) se localiza en el cromosoma 10q22-23, y codifica una proteína de igual nombre38 . Ésta es una fosfatasa lipídica, con actividad enzimática que primariamente remueve los grupos fosfatos de la tirosina, serina y treonina. Regula en forma negativa la vía del PI3K, inhibiendo la fosforilación del Akt, con actividad supresora tumoral. Esta proteína regula importantes funciones celulares tales como: crecimiento, proliferación, ciclo celular, apoptosis, migración e invasión celular y angiogénesis38-40 . En 1996 se identificó por primera vez una mutación germinal del PTEN, en un paciente con enfermedad de Cowden38 . Desde entonces y hasta la fecha se han detectado mutaciones en este gen en diferentes síndromes; en muchos casos con superposición clínica. Es debido a estos hallazgos que en la actualidad se considera, desde el punto de vista molecular, el concepto de síndrome ha-martomatoso-tumoral del PTEN, caracterizado por mutaciones en dicho gen y que se expresa clínicamente en un espectro de enfermedades relacionadas, entre las que se incluyen: enfermedad de Cowden, síndrome de Bannayan-Riley-Ruvalcaba, síndrome Proteus, síndrome símil Proteus41-44 .
Este concepto tiene un importante impacto clínico, dado que todas las entidades clínicas asociadas a mutaciones en el PTEN deben monitorizarse como si fueran una enfermedad de Cowden, dado el riesgo de cáncer órgano-específico que pueden desarrollar.
En esta actualización nos referiremos a dos de estas entidades que se asocian a poliposis.
ENFERMEDAD DE COWDEN O SÍNDROME DE LOS HAMARTOMAS MÚLTIPLESLa enfermedad de Cowden fue descrita, en 1963, por Lloyd y Denis45 , quienes la denominaron con el apellido de la paciente. En 1972, Weary et al46 consideran más adecuado el término síndrome de los hamartomas múltiples, dado que define mejor la asociación de estas displasias. Es una genodermatosis que presenta anomalías ectodérmicas, endodérmicas y mesodérmicas.
Se hereda en forma autosómica dominante, y afecta a ambos sexos, algo más frecuente en mujeres. Su incidencia es de 1 en 200.00040 .
No se sabe con certeza cuántos casos son esporádicos (debido al subdiagnóstico de la entidad), y se presume que un 40-65% son familiares (2 o más parientes afecta-dos)40 .
La enfermedad de Cowden está determinada por una mutación germinal en el gen PTEN. Aproximadamente el 61% de las mutaciones en este síndrome se localizan en los exones 5, 7 y 8, con el 38% de mutación en el exón 5 (que representa el 20% de la secuencia codificante). Existe una correlación fenotipo-genotipo, dado que mutaciones en el motivo fosfatasa 5’ están asociadas a deterioro multiorgánico41,47 . Existen en la bibliografía reportes de superposición en una misma familia de enfermedad de Cowden y síndrome de Bannayan-Riley-Ruvalcaba. Dicha asociación (como se dijo) se basa en que ambas entidades presentan mutaciones del PTEN42,43 .
Teniendo en cuenta el Consorcio Internacional del Síndrome de Cowden, se establecieron los criterios diagnósticos de la entidad (tabla III)39 .
Manifestaciones cutáneasLas alteraciones cutáneas son variadas; más de un 90% de los casos tienen manifestaciones a los 20 años; en el 99% de éstos, alrededor de los 29 años32 .
La facies es característica: se observa craneomegalia, facies adenoide, aspecto untuoso de la piel32 .

TABLA III. Criterios diagnósticos de la enfermedad de Cowden
Los triquilemomas faciales múltiples son un signo patognomónico, y preceden a las alteraciones internas. Se desarrollan a partir de la tercera década de la vida. Se presentan como pápulas, de 1 a 4 mm de diámetro, que se localizan preferentemente alrededor de la boca, la nariz y las orejas. Son color piel normal o ligeramente amarillo amarronadas, de superficie verrugosa, y pueden presentar un poro dilatado (fig. 7) 39 .
El fibroma de Cowden es una pápula o nódulo de mayor tamaño, que se localiza en la cara o el cuero cabelludo, de consistencia firme, blanquecina, asintomática. Su histopatología se caracteriza por la presencia de bandas de colágeno acelular lamelares con ocasionales células gigantes7,32 .
En dorso de manos, antebrazos y piernas desarrollan lesiones queratósicas que simulan verrugas. Su histología muestra hiperqueratosis con acantosis sin evidencia de coilocitosis celular10 .
En palmas y plantas hay una queratodermia punteada, con pápulas translúcidas que pueden tener en su centro una depresión cupuliforme (recuerdan a las queratodermias arsenicales). Su histología corresponde a una ortoqueratosis masiva, muy compacta en la capa córnea, con granulosis y acantosis10,32,39 .
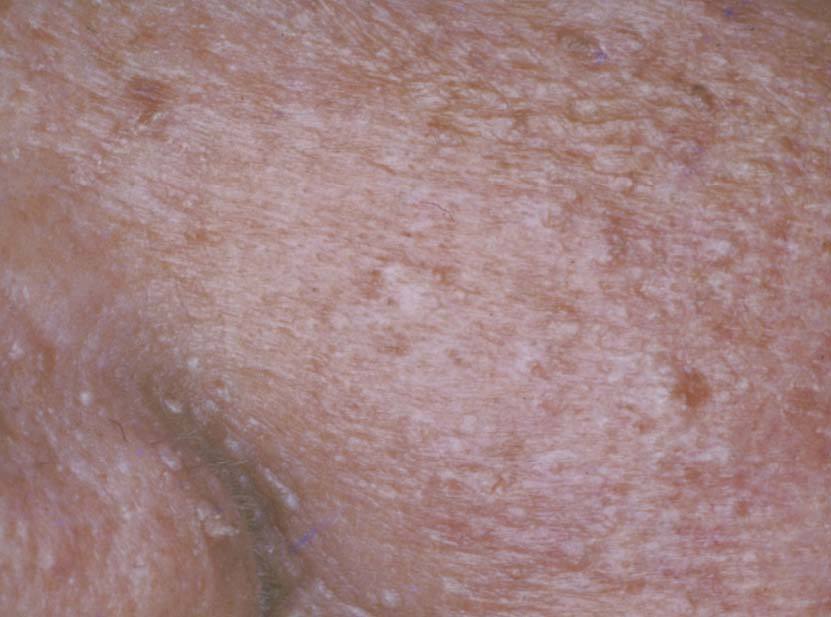
Figura 7. Triquilemomas múltiples en la cara. Enfermedad de Cowden.
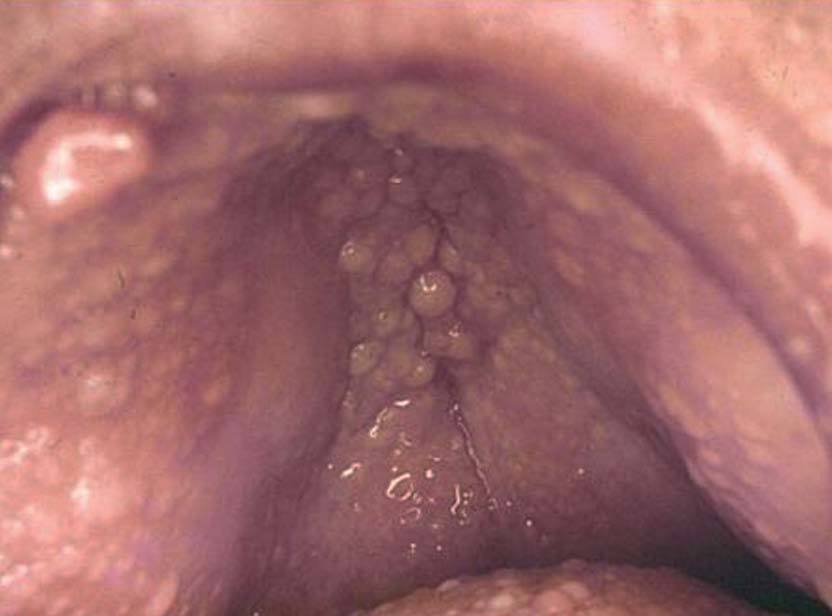
Figura 8. Empedrado del paladar. Enfermedad de Cowden.
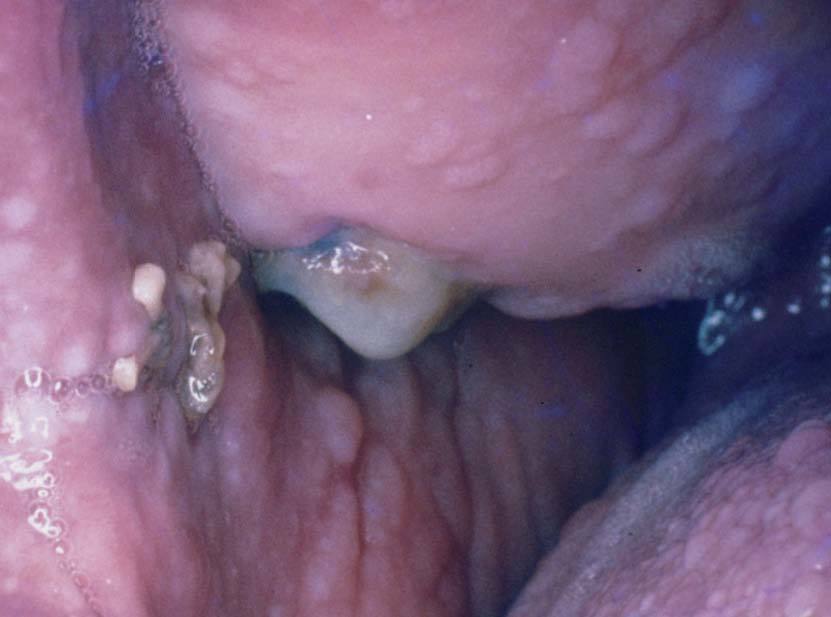
Figura 9. Pápulas en la mucosa yugal. Enfermedad de Cowden.
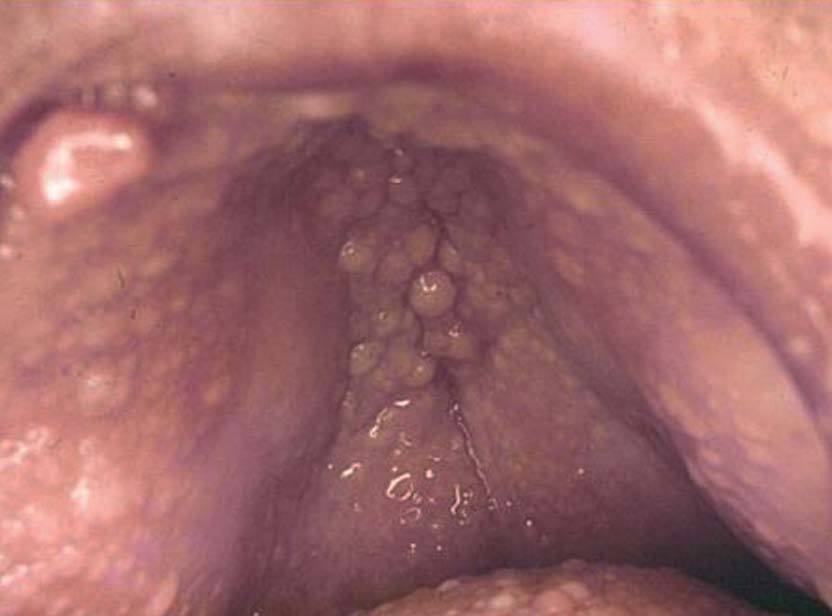
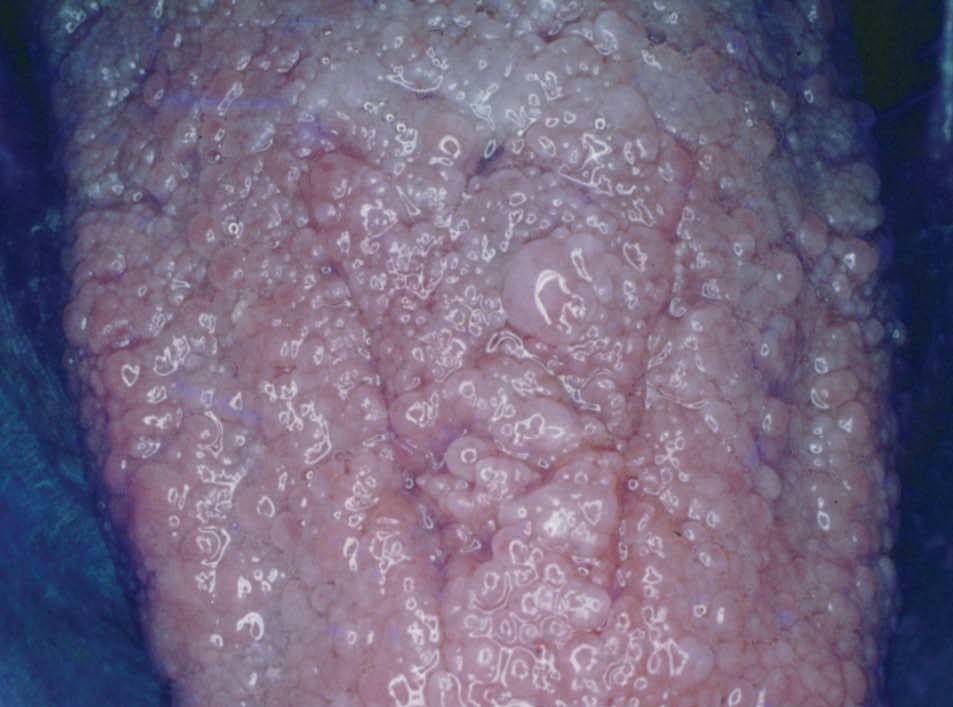
Las lesiones de mucosa oral son un hallazgo frecuente. Se evidencian pápulas de 1 a 3 mm de diámetro, de superficie lisa, del color de la mucosa normal o rosadas, que dan un aspecto de empedrado (fig. 8). Se localizan en toda la mucosa, incluidos paladar, lengua (figs. 9 y 10), labios y orofaringe. La histología muestra proyecciones fibrovasculares tapizadas por epitelio con o sin hiperplasia, según los sectores7,32,48 .
Se han reportado otras lesiones cutáneas: lipomas, angiolipomas, hemangiomas, poiquilodermia, manchas café con leche y fibromas escleróticos múltiples 10 .
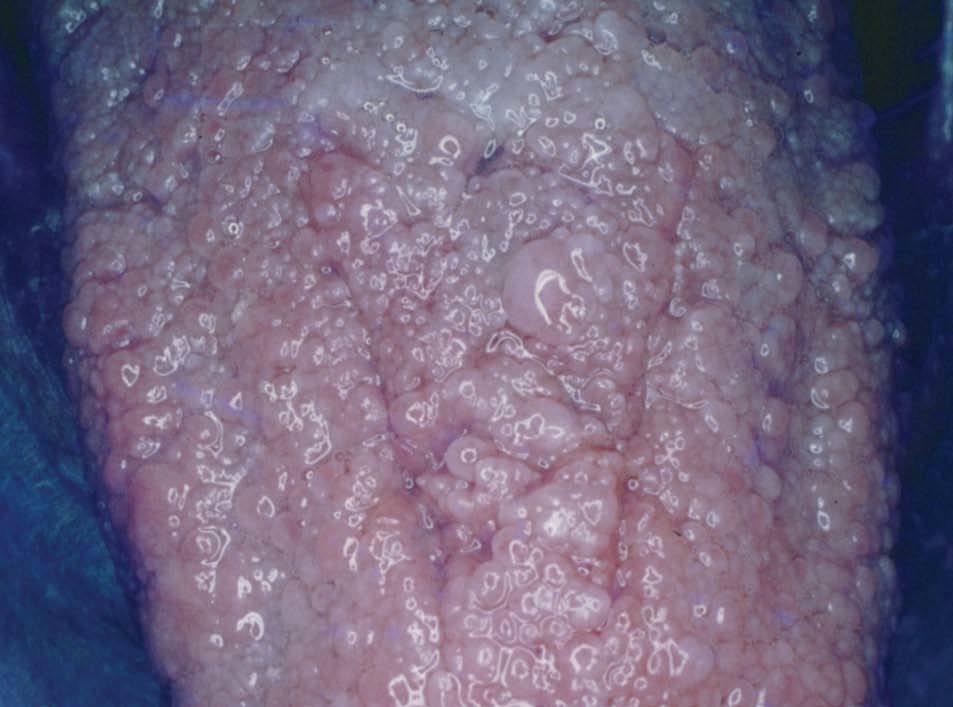
Figura 10. Pápulas en la lengua, imagen en empedrado. Enfermedad de Cowden.
Manifestaciones extracutáneasLas alteraciones tiroideas representan el hallazgo extracutáneo más frecuente, presente en el 62% de los casos. Afecta a ambos sexos, con diferentes tipos de lesiones, que incluyen bocio, adenomas benignos y quiste del conducto tirogloso. El cáncer de tiroides se observa con una frecuencia del 10% mayor que en la población general, con una histología predominante de carcinoma folicular. Se debe realizar controles periódicos para su detección temprana7,10,32 .
Las alteraciones mamarias están presentes en alto porcentaje. Se describe enfermedad fibroquística y fibroadenomas en el 76% de los casos. Se ha observado también papilomas ductales e hipertrofia virginal.
El carcinoma mamario se desarrolla en un porcentaje mayor que en la población general (25 a 50%), a una edad promedio de 38 a 45 años, y es bilateral en un 30% de los casos39,40 . En general, se corresponde histológicamente con carcinomas ductales y lobulares32 .
El cáncer de mama en el hombre es un componente menor del síndrome. Teniendo en cuenta este potencial maligno, se requieren exámenes mamográficos periódicos (cada 6 meses a 1 año), acompañados del autoexamen mensual, para la detección precoz de las lesiones7,10,48 .
En el tracto gastrointestinal, en el 85% de los pacientes se detectan múltiples pólipos, de tamaño menor a 5 mm; localizados a lo largo del tubo digestivo, desde el esófago hasta el ano, si bien son más frecuentes en el colon. Desde el punto de vista histológico, los pólipos son hamartomatosos, si bien pueden observarse también adenomas y ganglioneuromas. El potencial maligno de estos pólipos es bajo; debido a ello sólo se realizan estudios complementarios ante la presencia de síntomas39,48.
El tracto genitourinario está frecuentemente afectado en mujeres. Se destaca la presencia de quistes benignos de ovario y leiomiomas. Otras lesiones menos frecuentes incluyen teratomas, fibromas uterinos, pólipos de uretra, adenocarcinoma de uretra y cérvix, carcinoma renal y seminoma testicular 7,48,49 .
Entre las alteraciones esqueléticas también se han descrito: paladar alto y arqueado, cifosis, escoliosis, pectus excavatum, sindactilia, manos y pies grandes y alteraciones digitales. Las alteraciones oculares están presentes en un 13% de los casos e incluyen estrías angioides y miopía32,39,48 .
SÍNDROME DE BANNAYAN-RILEY-RUVALCABA (SBRR)Este nombre fue propuesto por Cohen en 1990 para unificar las diferentes denominaciones que recibió el síndrome: Riley-Smith (1960), Bannayan-Zonana (1971), Ruvalcaba-Myhre (1980)50-54 . Se transmite en forma autosómica dominante51 . Se han detectado mutaciones germinales en el PTEN en el 60% de los casos, y la mayoría se halla en los exones 6 y 9. No queda claro qué ocurre en el 40% restante, si bien podrían existir diferentes mecanismos de inactivación del PTEN o quizá otros genes involucrados39,40,55,56 .
Manifestaciones cutáneasComo manifestación cutánea destacan las pigmentaciones en el pene y la vulva. Se observan máculas marrones de pocos milímetros de diámetro. La histología de las máculas muestra hiperplasia lentiginosa de la epidermis y aumento de la melanina basal51,57 .
Otros componentes del síndrome incluyen: lipomas (tanto subcutáneos como viscerales), angiomas y acantosis nigricans51,57 .
Manifestaciones extracutáneasEl síndrome se caracteriza por la presencia de macrocefalia, con tamaño ventricular normal, retardo madurativo e hipotonía, que se evidencian en forma temprana. También se han observado deformaciones vasculares del sistema nervioso central51,52,57 .
Los pólipos se localizan primariamente en el colon y, desde el punto de vista histológico, son hamartomas, si bien en ciertos casos se han observado ganglioneuromas y ocasionalmente cambios adenomatosos57,58 .
También se describen alteraciones esqueléticas asociadas.
NEUROFIBROMATOSIS 1La neurofibromatosis 1 es el desorden neurogenético más frecuente, y afecta aproximadamente a 1 entre 2.500 y 3.300 nacidos vivos 58 . Se caracteriza por la presencia de manchas café con leche; efelidoides; nódulos de Lisch, múltiples neurofibromas y un variado espectro de alteraciones del sistema nervioso. Existen criterios bien establecidos para esta entidad 59 (tabla IV). Se hereda en forma autosómica dominante, si bien son frecuentes las mutaciones frescas. Tiene penetrancia completa y expresión variable, aun entre miembros de una familia 60 . El gen causante es el NF1 , que se localiza en el cromosoma 17q11.2, y su producto proteico se denomina neurofibromina, que está compuesta por 2.818 aminoáci- 61-63 . La secuencia de esta proteína revela cierta similitud con el dominio catalítico del Ras-GTPasa, con acción supresora tumoral. Las moléculas GAP (como la neurofibromina) inactivan el Ras, mediante señales mitogénicas, e inhiben la proliferación celular. La pérdida de expresión del gen NF1 resulta, en ausencia de función, neurofibromina Ras-GAP, hecho que determina aumento de actividad Ras, y da como resultado incremento de actividad celular y formación tumoral. Estudios recientes sugieren que esta actividad Ras-GAP no es la única función de la neurofibromina. Estudios en neuronas animales demostraron que tiene además actividad reguladora positiva sobre AMPc intracelular. Se observó que a través de esta vía regula el crecimiento de los astrositos; por lo tanto la pérdida de neurofibromina contribuye al incremento de la proliferación celular 64-66 . Se han identificado más de 500 mutaciones en este gen, en su mayoría exclusivas de cada grupo familiar. Un alto porcentaje de ellas determinan una versión extremadamente corta de neurofibromina 62 .
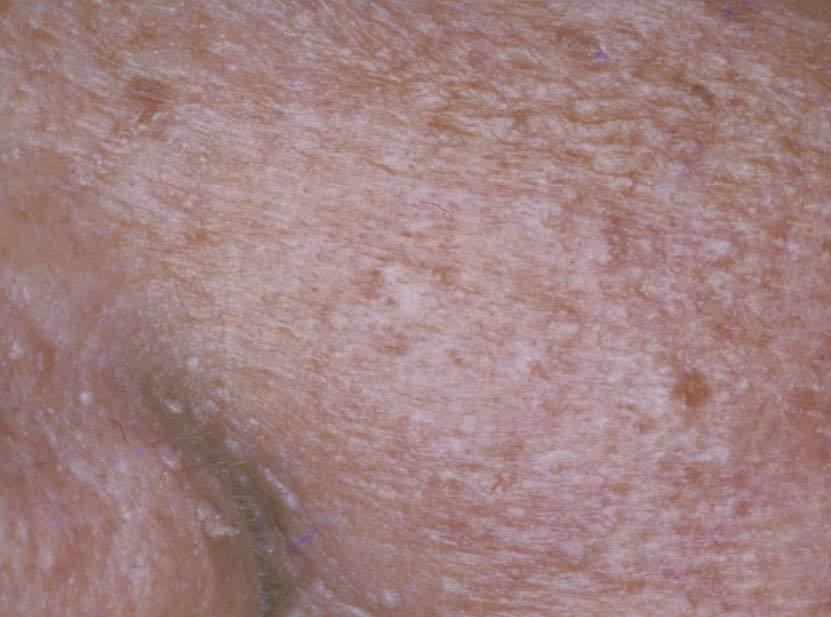
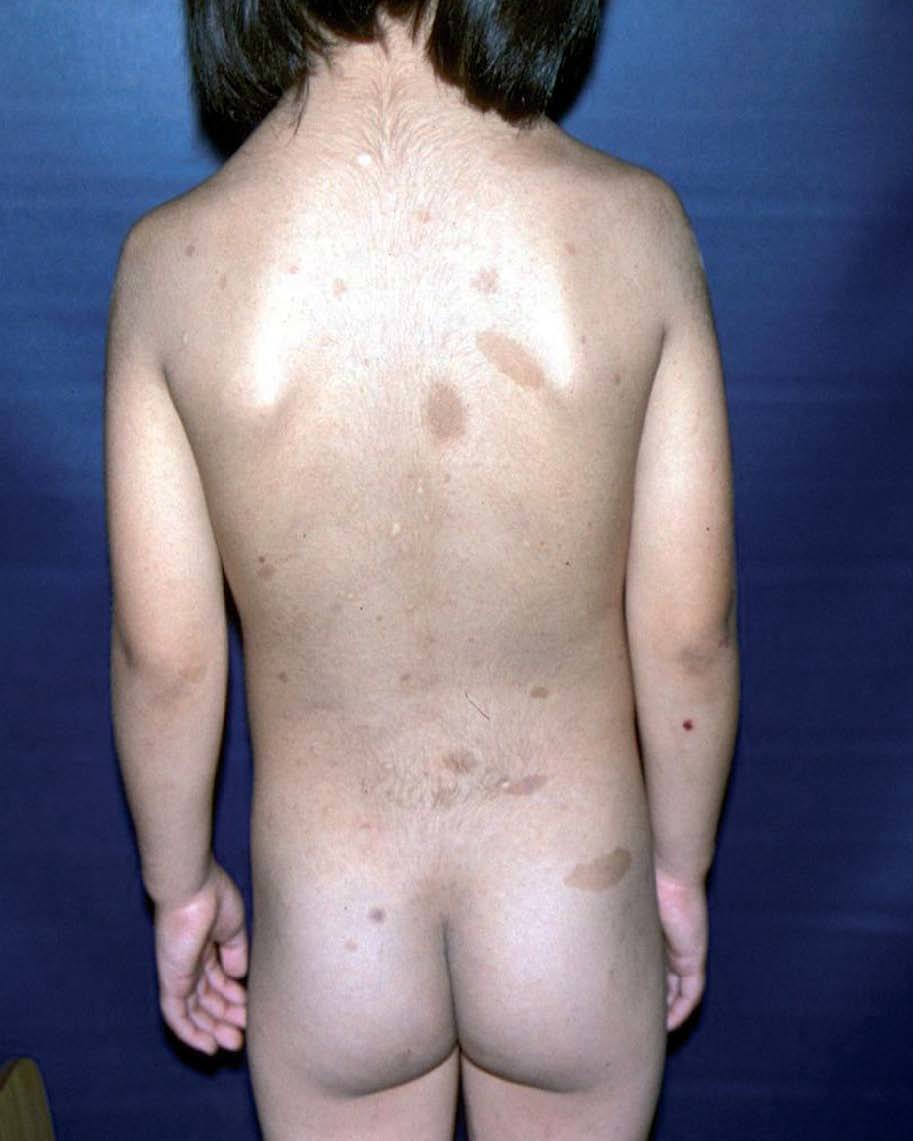
Manifestaciones cutáneasLas manchas café con leche (MCCL) a menudo se presentan en el primer año de vida; en la mayoría de los casos están bien establecidas a los 4 años de edad, y aumentan de número y tamaño con el tiempo. Se localizan en el tronco y las extremidades, de tamaño y forma variables. Son de color uniforme canela o pardo. La histología revela aumento de melanina en la capa basal. La presencia de más de 6 MCCL mayores a 0,5 cm en prepúberes, o mayores a 1,5 cm en adultos, es un criterio diagnóstico de la entidad59,67,68 (fig. 11).
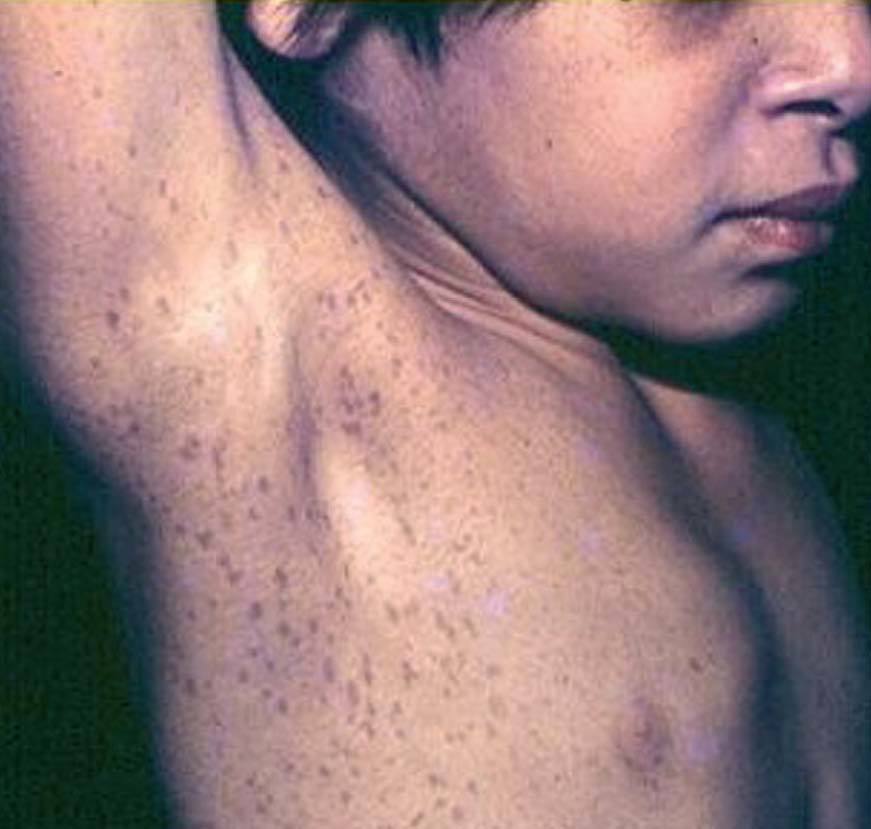
Las efelidoides están presentes en el 80% de los adultos con NF1 , si bien se ven con frecuencia en la infancia. Son pequeñas MCCL, semejantes a pecas (a diferencia de éstas se localizan tanto en área expuesta como no expuesta). Se distribuyen por todo el tegumento, y se consideran patognomónicas las ubicadas en la cúpula de la axila (signo de Crowe), también son significativas en el cuello y la ingle67 (fig. 12). Dentro de las MCCL existe una forma particular que está presente desde el nacimiento, es de gran tamaño, color más oscuro, bordes irregulares, y precede a la aparición de neurofibromas plexiformes, que se desarrollarán debajo de ella (se denomina mácula supraneurofibroma plexiforme)69 .
Con respecto a los tumores, se dividen en cutáneos y subcutáneos. Los primeros son los neurofibromas y son los más frecuentes. Son lesiones salientes, sésiles o pediculadas, de color piel normal o pigmentadas, blandas, y se invaginan al presionarlas (signo del ojal). Aparecen en la niñez y aumentan en número y tamaño con la edad69 .
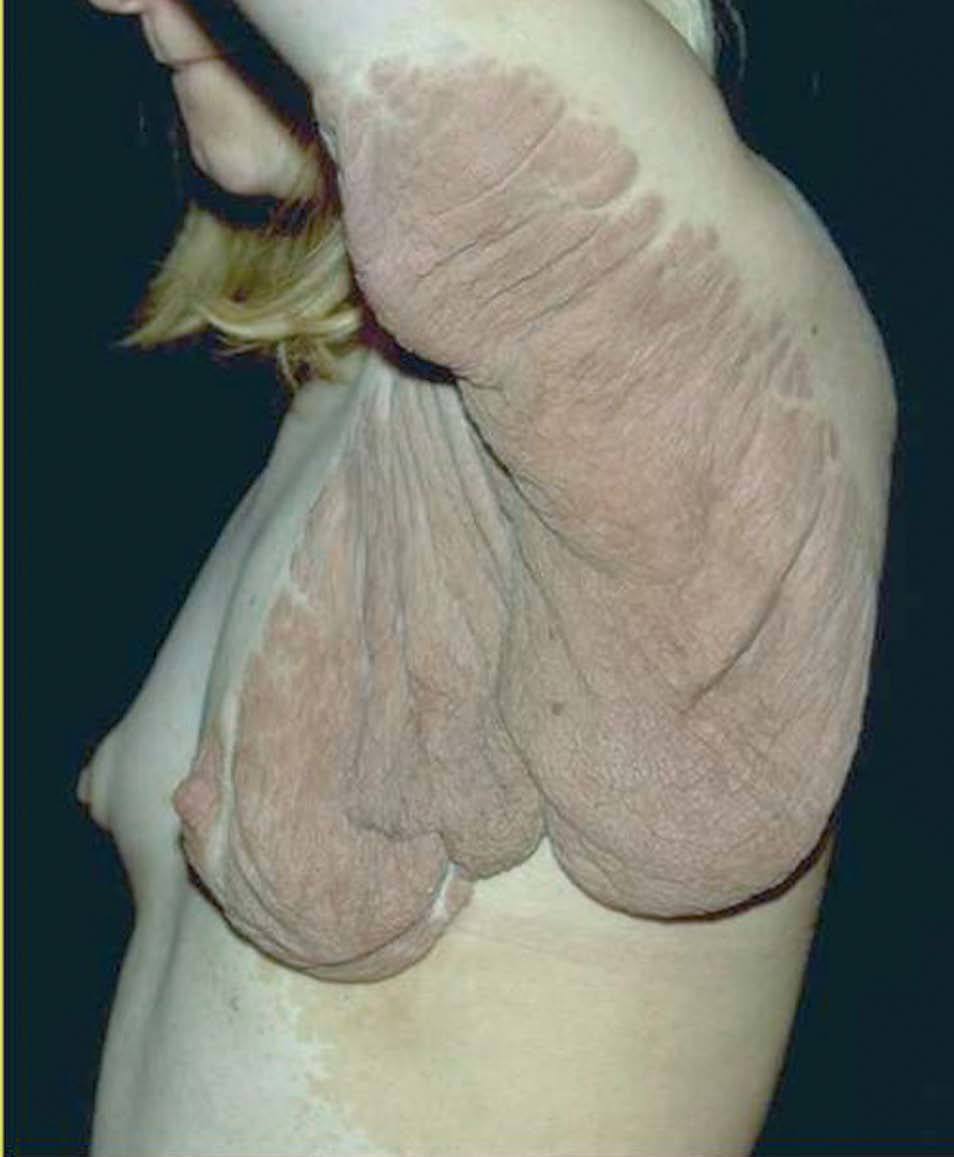
Entre los tumores subcutáneos se encuentran los neurofibromas plexiformes. Son de gran tamaño, y pueden deformar una región, la piel que los recubre puede ser hiperpigmentada o normal 69,70 (fig. 13).
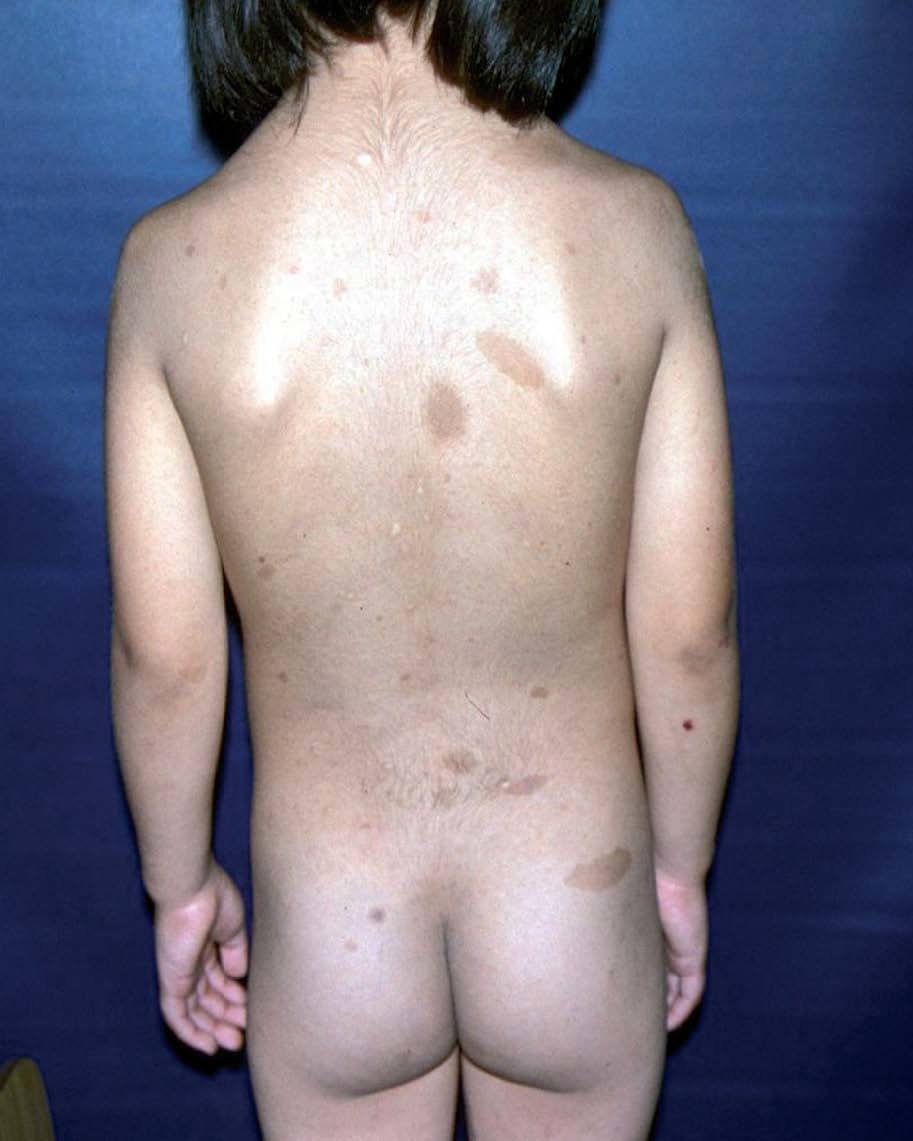
Figura 11. Manchas café con leche, de diversos tamaños, en número mayor a 6. NF1.
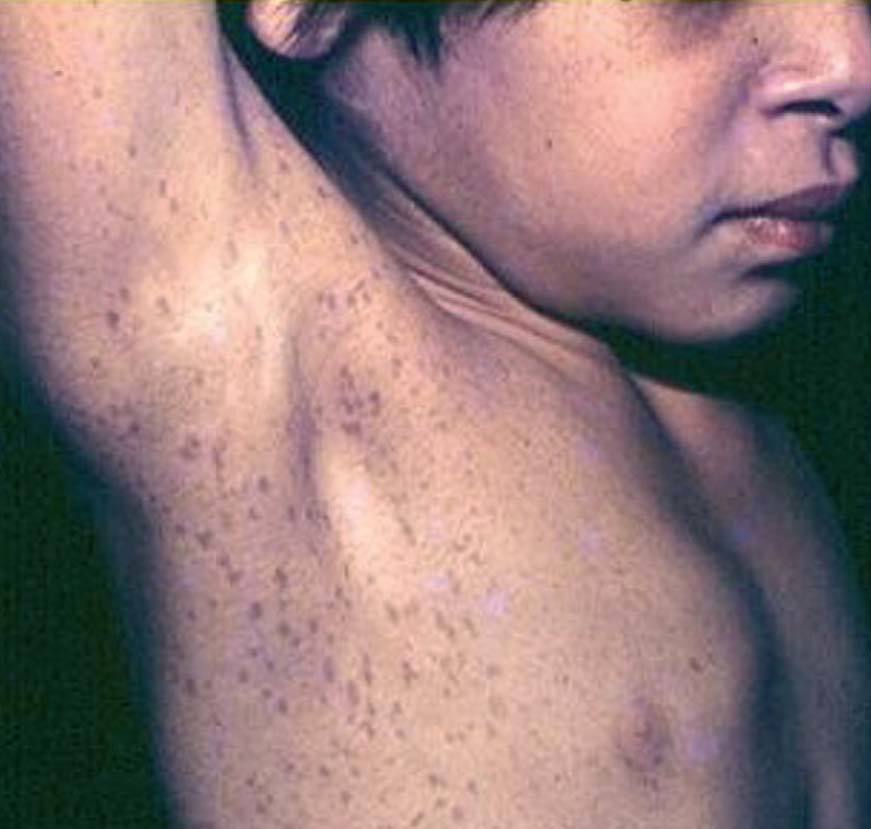
Figura 12. Signo de Crowe. NF1.

TABLA IV. Criterios diagnósticos de la neurofibromatosis 1
Se requieren por lo menos 2 de los siguientes signos: 6 o más MCCL de +5 mm en prepúberes; de +15 mm en adultos Al menos 2 neurofibromas o 1 neurofibroma plexiforme Efelidoides axilares o inguinales Nódulos de Lisch (más de 2) Glioma óptico Lesión ósea distintiva (seudoartrosis o displasia del efenoides) Familiar en primer grado con diagnóstico de NF1
Los neurinomas o schwannomas son nódulos pequeños, profundos, poco numerosos, dolorosos al tacto, con desplazamiento lateral70 .
Manifestaciones extracutáneasEn la región ocular presenta múltiples nódulos de Lisch (hamartomas melanocíticos del iris) que se visualizan con lámpara de hendidura. Su detección aumenta con la edad, a los 3 años, 5%; a los 6 años, 55%; pre-púbe-res, 94%, y en adultos, 97-100%71 .
El glioma del nervio óptico es el tumor que se observa con mayor frecuencia en la infancia; el 85% de los casos se producen antes de los 15 años. Corresponde, desde el punto de vista histológico, a un astrocitoma grado I o II. Puede provocar pérdida de la visión, alteraciones neuroendocrinas o hidrocefalia72 .
En el sistema nervioso se detectan diferentes tipos de tumores: astrocitomas, meningiomas, schwannomas73,74 . Por medio de resonancia magnética se observan imágenes hiperintensas, no específicas, más frecuente en ganglios basales y tálamo; su origen es controvertido70,74 .
En la región ósea, son criterios diagnósticos la seudoartrosis o la displasia del esfenoides. La cifosis y/o escoliosis y la escápula alada son otras de las alteraciones esqueléticas observadas con frecuencia59,60 .
La insuficiencia gastrointestinal se detecta en un 11-25% de los adultos afectados, y está determinada por la presencia de neurofibromas en la mucosa y submucosa, posiblemente a partir del plexo de Auerbach. Por medio de estudio de imágenes se observan formas polipoideas, localizadas a lo largo del tubo digestivo; los sitios más frecuentes son el estómago y el yeyuno; en raras ocasiones pueden detectarse en el esófago y el colon. Normalmente son lesiones asintomáticas, si bien pueden producir dolor abdominal, melena, constipación, obstrucción, perforación e intususcepción. Existen casos publicados de transformación maligna 75-77 . Se debe realizar seguimiento clínico del adulto con neurofibromatosis, y se harán estudios complementarios según la sintomatología 74 .
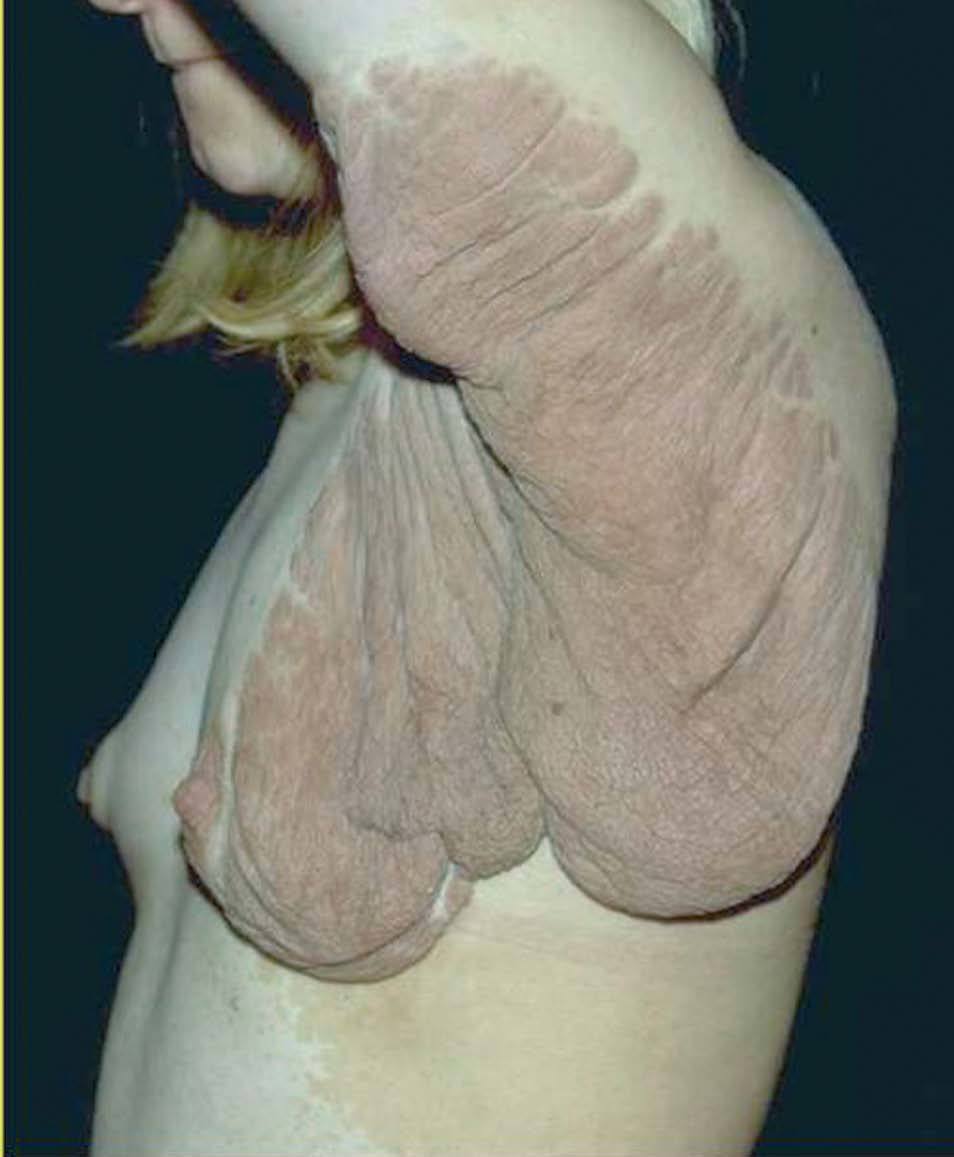
Figura 13. Neurofibroma plexiforme extensor. NF1.
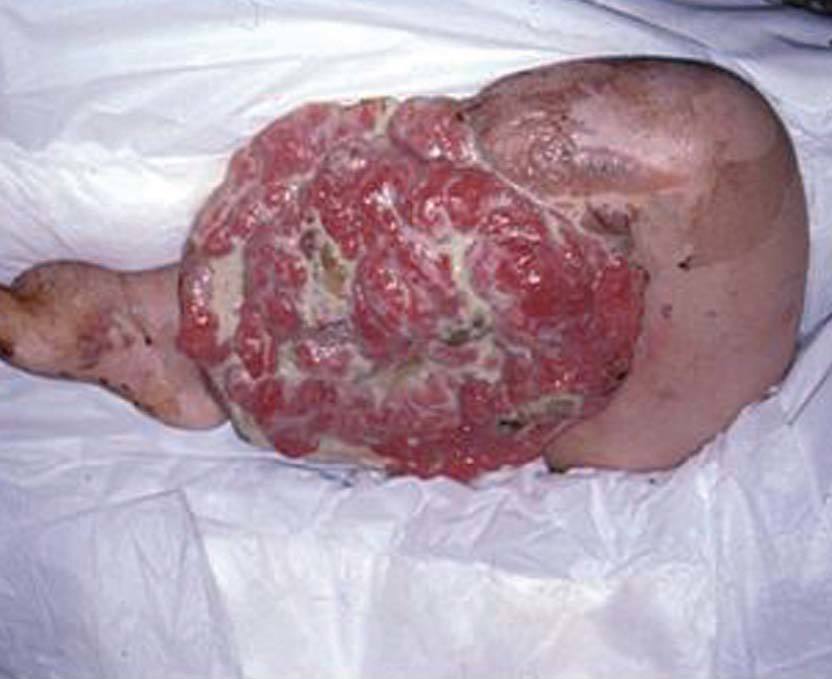
Figura 14. Fibrohistiocitoma maligno. NF1.
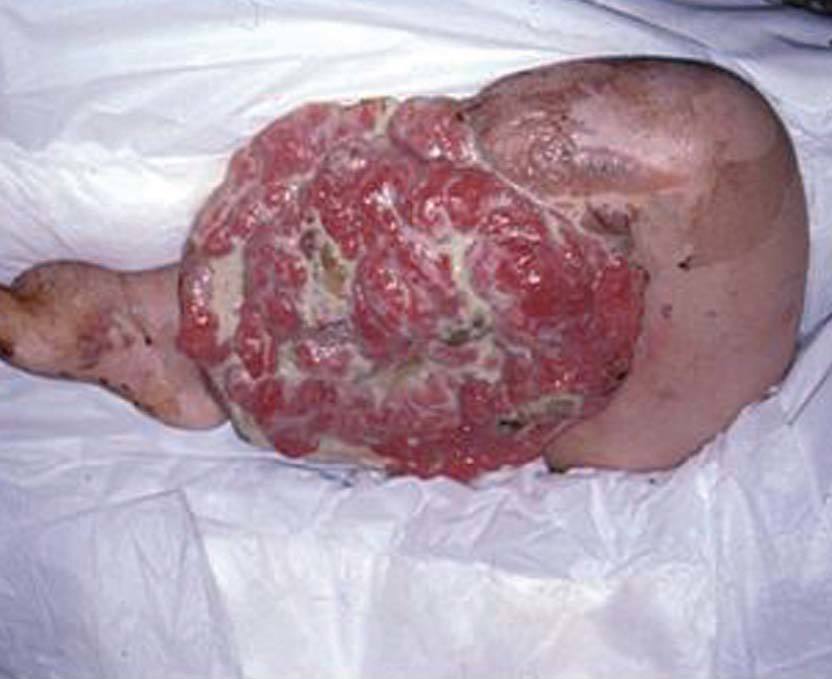
La incidencia de cáncer en pacientes con NF1 es un 15% mayor que en la población general. Debemos destacar el tumor maligno de vainas nerviosas periféricas, en un 3 al 5% de los casos (antes denominado schwannoma maligno, sarcoma neurogénico), puede desarrollarse de novo o a partir de un neurofibroma plexiforme 78 . El feocromocitoma se observa en adultos jóvenes con NF1, y se manifiesta clínicamente con hipertensión arterial 79 . Se refieren en la bibliografía con baja frecuencia liposarcomas y fibrohistiocitoma maligno (fig. 14), entre otros 80 .
Los pacientes con NF1 que asocian xantogranuloma juvenil tienen 221 veces mayor riesgo a padecer leucemia mielocítica crónica; 4 a 5 veces más leucemia linfoblástica aguda, y 10 veces mayor riesgo de linfoma no hodgkiniano81 .
SÍNDROME DE CRONKHITE CANADACronkhite y Canada, en 1952, describieron un síndrome al que le pusieron su nombre. Es una entidad rara, adquirida, no familiar, que aparece en forma consecutiva a estrés físico o emocional. Presenta alteraciones cutáneas asociadas a poliposis82 .
Se presenta con mayor frecuencia en varones, con una edad promedio de 59 años.
Manifestaciones cutáneasLas manifestaciones cutáneas preceden a las alteraciones intestinales. Se observan máculas hiperpigmentadas en parches, localizadas en brazos y manos, si bien pueden observarse en extremidades inferiores, cabeza, cuello y tronco en orden de frecuencia decreciente83-85 . El estudio histopatológico revela aumento de la pigmentación basal sin hiperplasia melanocítica.
Usualmente se desarrolla alopecia rápidamente progresiva, en pocas semanas, e involucra áreas de pelo terminal y provoca alopecia difusa parcial o total. El mecanismo de producción es desconocido84-86 .
Las uñas se ven afectadas. Los cambios incluyen onicolisis de inicio proximal y puede progresar a onicomadesis. Se ha observado también: onicosquizia, onicodistrofia y un patrón característico que se presenta como un triángulo invertido de uña con distrofia de la lámina82,85,86 .
La piel, el pelo y las uñas tienen una apariencia cérea. El curso de estas alteraciones es independiente de la poliposis, y se resuelve espontáneamente84-86 .
Manifestaciones extracutáneasSe caracterizan por la presencia de poliposis intestinal difusa, de tipo inflamatorio o de retención (no neoplásico); si bien se han descrito adenomas en raras ocasiones. A pesar de su naturaleza benigna, se asocian a alta mortalidad debido al deterioro del estado general de estos pacientes.
Desarrollan cuadros de diarrea, asociados a pérdida de peso, dolor abdominal y otras complicaciones gastrointestinales, como enteropatía perdedora de proteínas y desnutrición84,87 .
En el 15% de los casos se detecta adenocarcinoma gástrico o colónico. Si bien estos pacientes tienen una edad en la cual la malignidad gastrointestinal es frecuente, el proceso inflamatorio crónico de la mucosa propia podría predisponer a la neoplasia. Sin embargo, hasta el presente se desconoce la etiología del síndrome y su relación causal con la neoplasia 87 .
Correspondencia: Dra. P. Della Giovanna. Italia 1151. San Miguel (1663). Buenos Aires. Argentina. Correo electrónico: dellagiovannap@hotmail.com