





El objetivo del presente estudio es describir el perfil epidemiológico de los pacientes con síndrome de Sweet así como las características etiopatogénicas, evolutivas y terapéuticas, y comparar los resultados con estudios previamente realizados.
Hemos realizado un estudio descriptivo retrospectivo de los pacientes valorados por el Servicio de Dermatología del Hospital General Universitario Gregorio Marañón de Madrid entre enero de 1990 y enero de 2009, con juicio clínico de síndrome de Sweet y confirmación histopatológica.
Tras analizar los resultados de los 21 casos reunidos y comparar con estudios previos, concluimos que el síndrome de Sweet es una entidad infrecuente pero no rara, que aparece predominantemente en sujetos de mediana edad, con localización preferente en las extremidades. La biopsia cutánea es una herramienta diagnóstica muy rentable.
Aunque la mayoría de los casos son idiopáticos debe descartarse cuidadosamente la posibilidad de que se trate de un proceso paraneoplásico o asociado a una enfermedad sistémica. Debe prestarse especial atención a la detección sistemática de procesos hemoproliferativos. Por último, hay que valorar la posibilidad de que se trate de un síndrome de Sweet inducido por fármacos. El tratamiento de elección son los corticoides sistémicos, con tan buena respuesta que constituye un criterio diagnóstico. El yoduro potásico puede considerarse como un tratamiento de primera línea dada su efectividad, similar a los corticoides, y con menores efectos adversos, sobre todo en el tratamiento de las recurrencias.
The aim of this study is to describe the epidemiological profile of patients with Sweet's syndrome and the aetiopathogenic, developmental and therapeutic features and to compare the results with previous studies.
We conducted a retrospective, descriptive study of patients evaluated by the department of Dermatology of Hospital General Universitario Gregorio Marañón in Madrid, between January 1990 and January 2009 with a clinical diagnosis of Sweet's syndrome and histopathological confirmation.
After analysing the results of the 21 cases collected and comparing them with previous studies, we conclude that Sweet's syndrome is an uncommon but not rare disease that occurs predominantly in middle-aged subjects, with preferential localised in the extremities. Skin biopsy is a very cost effective diagnostic tool. Although most cases are idiopathic, the possibility of para-neoplastic process or associated systemic diseases must be carefully excluded. Special attention must be paid to screening of haemoproliferative processes. Finally, the possibility of drug-induced Sweet's syndrome should be taken into account.
The treatment of choice is systemic corticosteroids, with such a good response that constitutes a diagnostic criterion. Potassium iodide can be considered a first line treatment, given its effectiveness, similar to steroids, and with fewer side effects, especially in the treatment of recurrences.
El síndrome de Sweet, o dermatosis neutrofílica febril aguda, se trata de una enfermedad infrecuente. En la literatura médica, la mayoría de las descripciones corresponden a casos aislados o a series cortas de casos cuyo número oscila entre los 2–15 casos. La serie más larga, publicada por dermatólogos de la Clínica Mayo de Rochester, Estados Unidos, data de 1995 y reunió a 48 pacientes en el período comprendido entre 1980–1992. En nuestro medio no disponemos de series similares y desconocemos si en la actualidad las características clínicas e histopatológicas de los pacientes diagnosticados de síndrome de Sweet, así como las etiopatogénicas, evolutivas y terapéuticas, se superponen a las clásicamente descritas. Esto es de especial importancia con respecto a la posibilidad de enfermedades sistémicas asociadas o a su aparición como un proceso paraneoplásico. La problemática planteada nos llevó a revisar los casos de síndrome de Sweet, confirmados histopatológicamente, valorados en nuestro servicio de dermatología durante un período de 19 años.
Material y métodosHemos realizado un estudio descriptivo retrospectivo de los pacientes valorados por el Servicio de Dermatología del Hospital General Universitario Gregorio Marañón de Madrid entre enero de 1990 y enero de 2009, con juicio clínico de síndrome de Sweet y confirmación histopatológica. Todos los pacientes cumplían los criterios de Su y Liu (tabla 1) necesarios para el diagnóstico (2 mayores y 2 menores). Los datos obtenidos tras la revisión minuciosa de las historias clínicas se han analizado con el programa estadístico SPSS 11. Posteriormente, se ha realizado una revisión de la literatura médica.
Criterios diagnósticos del síndrome de Sweet
| Criterios mayores |
|
| Criterios menores |
|
| Diagnóstico: Se requieren 2 criterios mayores y 2 menores |
Se recogió un total de 21 casos de síndrome de Sweet con confirmación histopatológica, de los que 9 eran mujeres y 12 eran varones (57,1%), con una media de edad de 49 años. El 57% de los pacientes (12 de los 21) asociaba fiebre, con una temperatura media de 38,6°C, la mitad de ellos de forma previa a la aparición de las lesiones cutáneas y la otra mitad de modo simultáneo. Once de ellos referían, además, un cuadro pseudogripal asociado (malestar general, artromialgias y sensación distérmica).
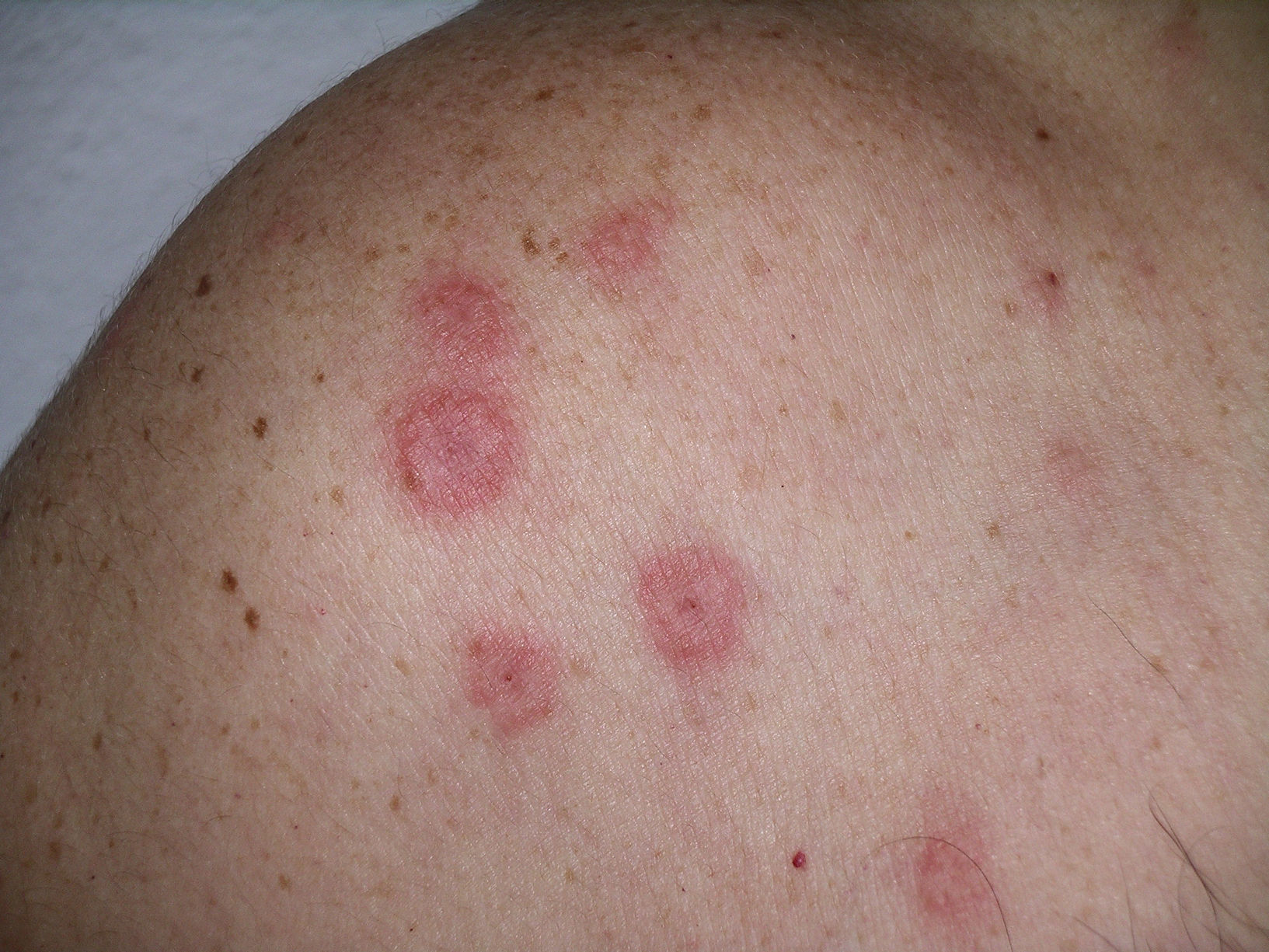
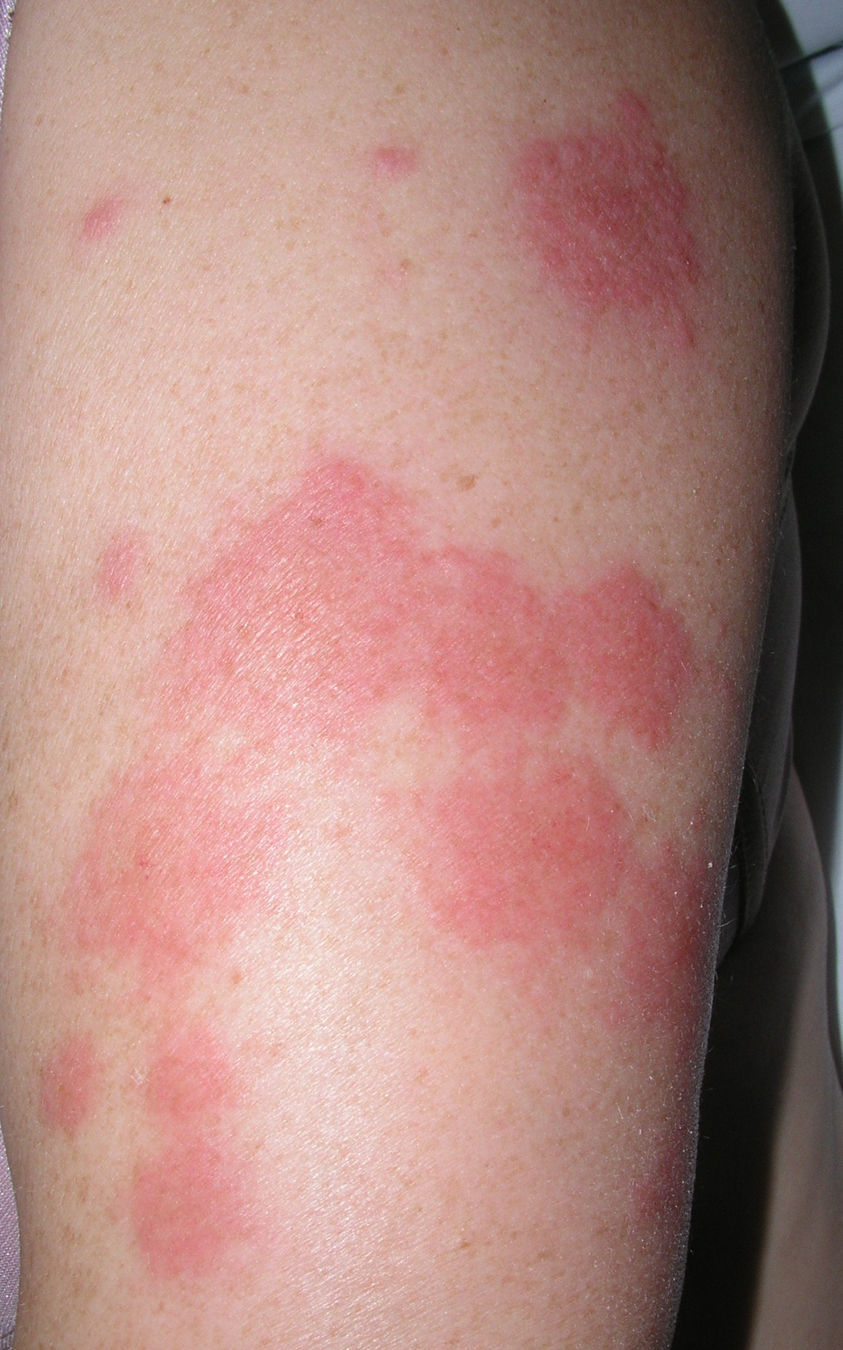
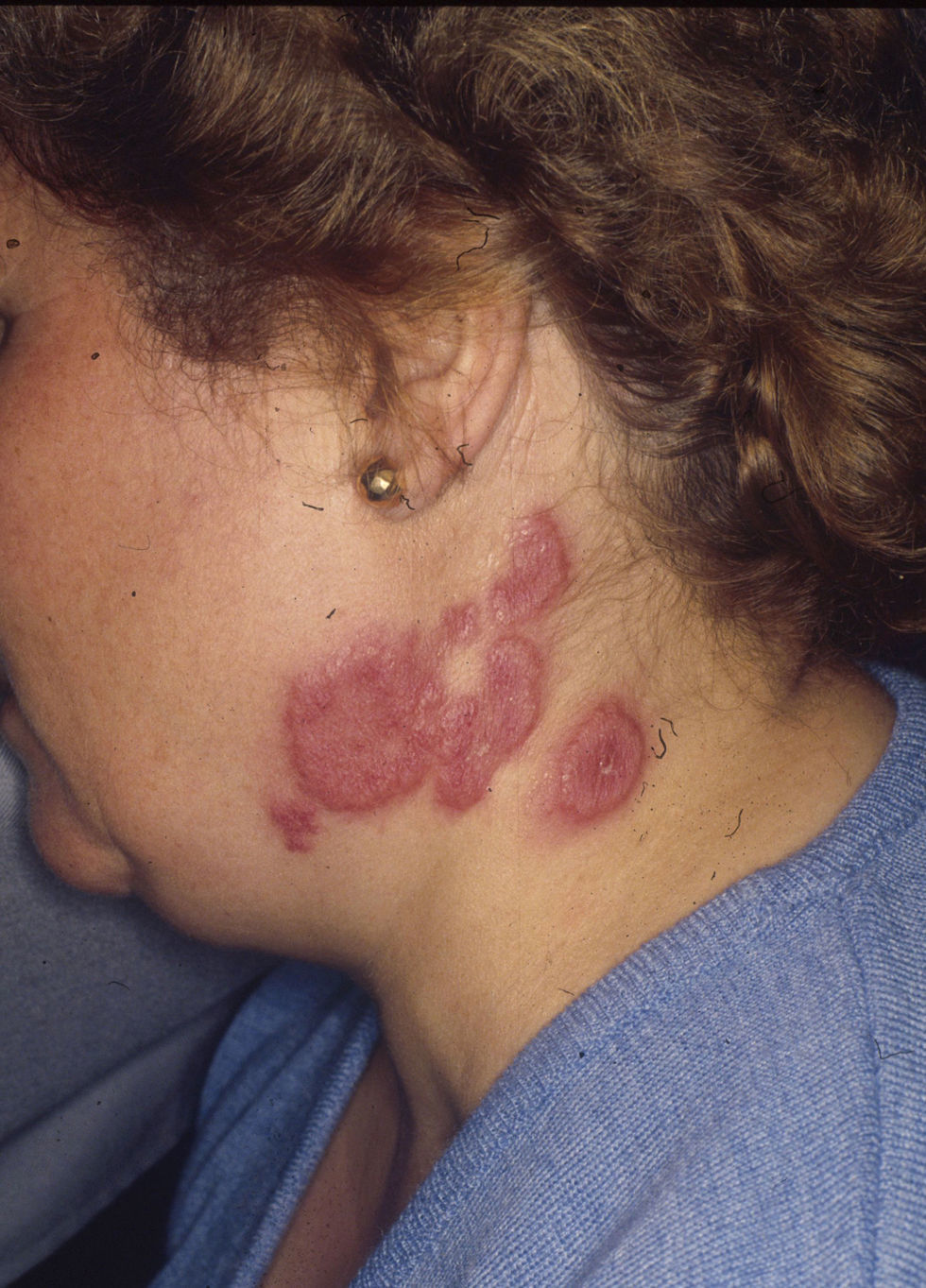
Las lesiones cutáneas se describieron como pápulas y placas eritematoedematosas en todos los pacientes (figs. 1–3). Aparecieron como lesión elemental única en 13 de los 20 pacientes, con aparición concomitante de nódulos en 5 pacientes y con aparición de vesiculoampollas en 3 pacientes. La distribución de las lesiones fue simétrica en la mayoría de los pacientes (17 de 21; 81%). Para valorar la localización de las lesiones se distinguieron las siguientes 6 áreas corporales: la cabeza, el cuello, el tórax, el abdomen, los miembros superiores y los miembros inferiores. En la mayoría de los pacientes (15 de 21) había afectación de más de un área corporal de las descritas. Las localizaciones más frecuentes fueron las extremidades (13 de 21 en los miembros superiores, y 13 de 21 en los miembros inferiores), seguidas de las lesiones a nivel del tórax (12 de 21) y de la cabeza (10 de 21). Las áreas afectadas con menor frecuencia fueron el abdomen (4 de 21) y el cuello (6 de 21). Respecto a los casos localizados en una sola área corporal (6 de 21), en 2 de ellos las lesiones se localizaban en la cabeza, en otros 2 casos se localizaban en los miembros inferiores y en los 2 casos restantes se localizaban en el tórax y los miembros superiores.



Todos los pacientes referían un inicio brusco de las lesiones y aproximadamente la mitad de ellos (10 de 21) referían las lesiones como dolorosas, 5 las referían como pruriginosas y los 3 restantes las referían como asintomáticas. Sólo en 3 de los 21 pacientes se describe afectación de las mucosas: conjuntival en 2 de ellos (epiescleritis) y oral en el tercero (ulceración aftosa concomitante). No se demostró afectación de otros órganos en ningún paciente.
En cuanto a las asociaciones con otras enfermedades, el 71,4% de los casos (15 de 21) fueron idiopáticos. Dos pacientes presentaron un cuadro infeccioso previo (infección pulmonar consolidativa y otitis media aguda). En 2 casos existía una enfermedad sistémica de base: espondilitis anquilosante en uno de ellos y en el otro las lesiones aparecieron en el contexto de un brote de colitis ulcerosa (CU). Por último, encontramos un cuadro hemoproliferativo asociado en 2 pacientes: policitemia vera y un síndrome mieloproliferativo concomitante, que a los 5 años desarrolló una leucemia aguda mielogénica.
En relación con el tratamiento, la mayoría de los pacientes recibieron corticoides por vía oral (12 de 21), 3 recibieron yoduro potásico, 3 recibieron colchicina y 3 no recibieron tratamiento alguno. De los que recibieron tratamiento con corticoides sistémicos, la mayoría recibieron prednisona en dosis de 0,5mg/kg, en pauta descendente (11 de 21), y uno recibió deflazacort en dosis equivalentes, con excelente respuesta en todos los casos.
Las pruebas complementarias analíticas demostraron anemia en 3 de los 21 pacientes y leucocitosis con neutrofilia en 10 de los 21 (47,6%). Entre los reactantes de fase aguda, el 57% (12 de 21) mostró elevación de la proteína C reactiva, el 33,3% (7 de 21) mostró elevación de la velocidad de sedimentación globular y en 2 pacientes del total se halló una trombocitosis. En 4 de los 21 pacientes se detectó elevación de los anticuerpos antiestreptolisina. La bioquímica y el sedimento urinarios de 3 de los 21 pacientes revelaron hematuria leve y en 2 de ellos revelaron proteinuria asociada. En 9 de los 21 pacientes se realizó radiografía de tórax, sin hallazgos relevantes en todos, salvo en un caso en el que se apreciaba un infiltrado previo en resolución. En 2 pacientes se detectaron anticuerpos antinucleares, y el factor reumatoide fue negativo en todos los pacientes. En el 61,9% (14 de 21) se solicitó un proteinograma, que fue normal en todos ellos.
En el 42,8% de los pacientes (9 de 21) hubo recurrencias de las lesiones cutáneas tras la suspensión del tratamiento (corticoide oral en todos ellos), con una mediana de 2 recurrencias. En el manejo de las recurrencias, los tratamientos más utilizados fueron los corticoides orales (4 de 9) y el yoduro potásico en dosis de 300mg cada 8h (4 de 9); en un caso (1 de 9) se pautó colchicina, con muy buena respuesta en todos ellos. Una de las pacientes desarrolló un síndrome de Sweet crónico, y las lesiones cutáneas recidivaron al suspender el tratamiento, por lo que precisó tratamiento continuado, que actualmente se lleva a cabo con colchicina.
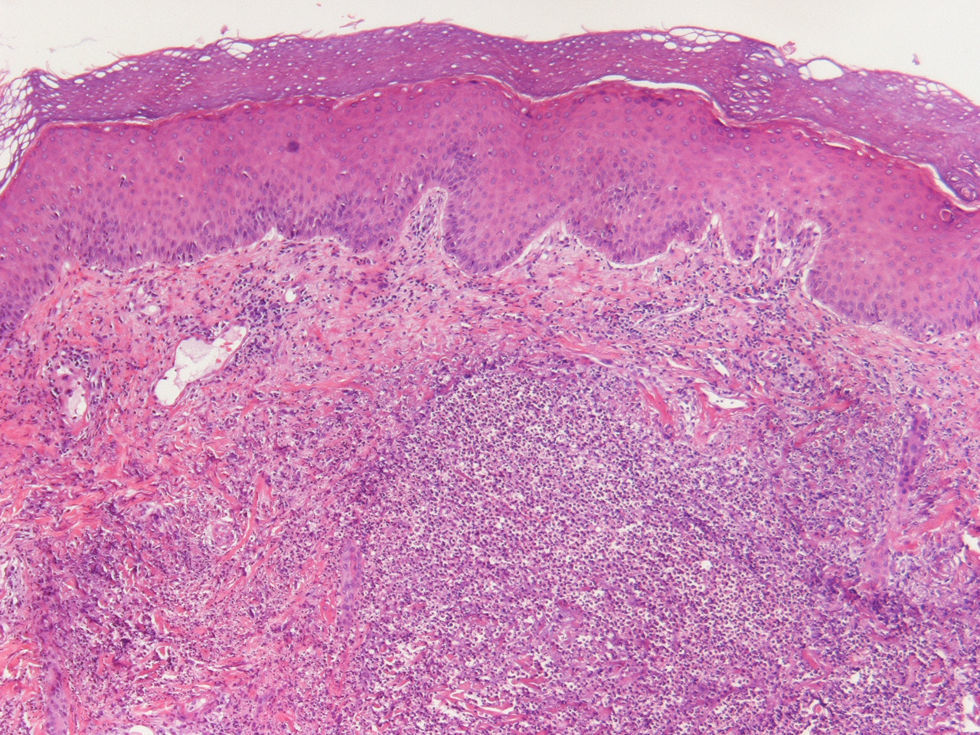
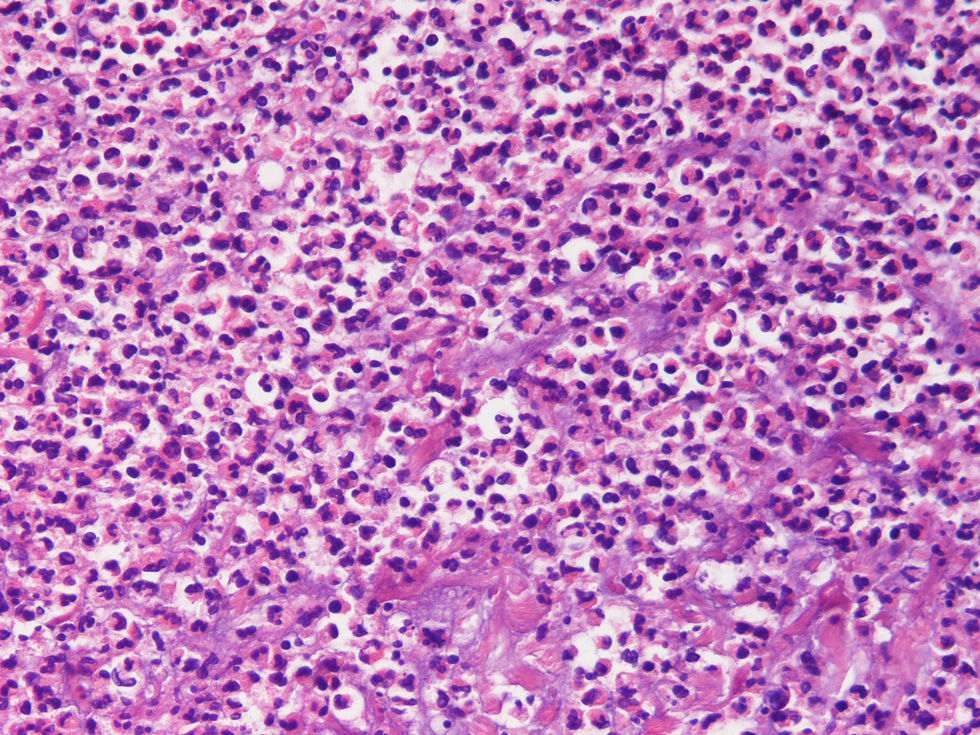
Dado que hemos considerado la confirmación histopatológica del síndrome de Sweet como criterio de inclusión obligado, todos los casos presentan una histología compatible con este cuadro. En el 100% de las biopsias cutáneas aparecen infiltrados dérmicos de polimorfonucleares neutrófilos, que en uno de los casos llega hasta la hipodermis. En el 90,5% (19 de 21) no aparecen cambios a nivel epidérmico. En el 100% de los casos existe ausencia de vasculitis leucocitoclástica asociada (figs. 4 y 5).
: la dermis se halla ocupada por un denso infiltrado inflamatorio de leucocitos polimorfonucleares neutrófilos. La epidermis aparece conservada.")

Detalle del infiltrado inflamatorio de la figura 4: se aprecia que el infiltrado inflamatorio está constituido mayoritariamente por neutrófilos, que muestran fragmentación nuclear.
En 1964, Robert Douglas Sweet describió 8 casos de mujeres de mediana edad que presentaban un cuadro de fiebre de inicio brusco, acompañado de placas eritematosas precedidas de cuadros de infección respiratoria o gastrointestinal inespecífica, y mostraban un infiltrado de polimorfonucleares neutrófilos en la biopsia cutánea. En 1968, Whittle informó de un caso similar y lo denominó síndrome de Sweet1,2.
Desde el punto de vista epidemiológico, el síndrome de Sweet, o dermatosis neutrofílica febril aguda, se trata de una enfermedad infrecuente, sin claro predominio racial, con discreta mayor incidencia en Japón, donde se ha descrito la asociación con HLA Bw54 y CW1. Se ha informado en la literatura médica de aproximadamente 500 casos. Existe un claro predominio femenino (2–4:1), con una edad media de inicio de 30–60 años. Hasta el 20% de los casos se considera paraneoplásico, sin diferencia entre sexos, mientras que los casos inducidos por fármacos son más frecuentes en el sexo femenino3,4.
La etiopatogenia no ha sido aclarada, aunque se ha relacionado con mecanismos de hipersensibilidad a antígenos bacterianos, virales o tumorales, así como con una alteración en la regulación local o sistémica de la secreción de citoquinas, sobre todo las de tipo 1 de los linfocitos T colaboradores, como la interleuquina 1 y el interferón γ5,6. Se distinguen 3 subtipos de síndrome de Sweet, según el proceso etiológico relacionado. Se han descrito casos familiares7. Otros trastornos neutrofílicos reactivos, como la hidradenitis neutrofílica ecrina, están estrechamente relacionados y podrían representar un espectro similar. El síndrome de Sweet parece estar encuadrado junto con otras entidades similares en el espectro de las dermatosis neutrofílicas8.
A nivel cutáneo, el síndrome de Sweet se caracteriza por placas o pápulas eritematosas, dolorosas, rara vez pruriginosas, edematosas, pseudovesiculosas, con una decoloración amarillenta central, que en ocasiones se hacen pseudopustulosas, vesiculosas, ampollosas e incluso llegan a ulcerarse. Además, en ocasiones se observan nódulos inflamatorios asociados, similares al eritema nodoso1,5. Se han descrito fenómenos de hipersensibilidad cutánea y las lesiones se localizan en sitios previamente estimulados (biopsias, arañazos de gato, picaduras de insectos, venopunción, catéteres intravenosos), con frecuentes recurrencias en sitios afectados de forma previa. Las localizaciones más frecuentes son la cara, el cuello y las extremidades superiores1. Los casos paraneoplásicos se caracterizan por una distribución variable y asimétrica, y las lesiones vesiculoampollosas y la ulceración son más frecuentes que en los casos idiopáticos. Las lesiones nodulares, similares al eritema nodoso, tienden a localizarse en los miembros inferiores9–11.
Las lesiones cutáneas tienden a desaparecer espontáneamente en 5–12 semanas sin dejar cicatriz; aparecen recurrencias hasta en el 30% de los pacientes, con o sin tratamiento, y hasta en el 50% de los casos asociados a trastornos hematológicos.
Hasta el 80% de los casos presenta fiebre. Los pacientes suelen referir un cuadro pseudogripal asociado o un cuadro infeccioso, generalmente una infección de vías respiratorias superiores, sobre todo por Streptococcus, seguida en frecuencia por cuadros gastrointestinales por Yersinia. Con frecuencia se aprecia epiescleritis asociada, que puede ser la manifestación inicial13. La malignidad hematológica se describe hasta en el 20% de casos, sobre todo la leucemia mielogénica aguda. La asociación con neoplasias sólidas es menor, y destacan las del tracto genitourinario, la mama y el colon11. Multitud de fármacos se han relacionado con el desarrollo del síndrome de Sweet, sobre todo con el factor estimulante de colonias de granulocitos13–16.
El diagnóstico se basa en el cumplimiento de ciertos criterios establecidos (Su y Liu 1986). Se entienden como lesiones típicas las placas o los nódulos dolorosos o sensibles, con vesículas, pústulas o ampollas, y se entiende como histología compatible la infiltración dérmica por neutrófilos, con ausencia de signos de vasculitis leucocitoclástica (tabla 1).
Los hallazgos histopatológicos demuestran una epidermis generalmente sin cambios, aunque pueden encontrarse espongiosis, vesículas intraepidérmicas o subepidérmicas, abscesos subcórneos o infiltración de los anejos por neutrófilos. A nivel dérmico suele apreciarse un infiltrado neutrofílico nodular, difuso y perivascular, sin evidencia de vasculitis, con ocasional leucocitoclasia sin necrosis fibrinoide. En la hipodermis puede observarse una paniculitis septal o lobulillar en las lesiones concomitantes de eritema nodoso y pioderma gangrenoso1,5.
Con la microscopia electrónica no se aprecian lesiones a nivel del endotelio vascular, sino signos de activación metabólica (abundancia de mitocondrias, retículo endoplásmico y múltiples complejos de Golgi). Los vasos se ven separados de los infiltrados celulares por una lámina electrolúcida, que refleja los fenómenos de reparación endotelial asociados. La inmunofluorescencia directa puede demostrar depósitos de inmunoglobulinas (Ig) (IgG, IgM), complemento (C3) y fibrina de forma difusa alrededor de vasos5.
En cuanto al tratamiento, los antibióticos en general son ineficaces y solo en el caso de infección específica sí mejoran el cuadro. Se han descrito casos de desaparición de lesiones de síndrome de Sweet en pacientes con enfermedad de Crohn tratados con metronidazol. Para los corticoides orales (prednisona de 0,5–1mg/kg/día), 4–6 semanas se considera el tratamiento de elección, y con ellos se obtiene tan buena respuesta que constituyen uno de los criterios diagnósticos del síndrome de Sweet. Puede prolongarse este tratamiento en dosis bajas 2 o 3 meses adicionales para suprimir las recurrencias. Si hay pocas lesiones localizadas, se pueden tratar con corticoides superpotentes tópicos o intralesionales. Los inhibidores de la calcineurina tópicos pueden ser útiles. Las alternativas terapéuticas incluyen yoduro potásico (900mg/día), dapsona (100–200mg/día), colchicina (1,5mg/día), antiinflamatorios no esteroideos (indometacina, naproxeno), ciclofosfamida, ciclosporina, clofacimina, interferón α y sulfapiridina5,6,10–19.
Tras revisar la literatura médica hemos encontrado algunas series de pacientes con síndrome de Sweet, la mayoría con menos de 15 pacientes20–24. La serie más larga que hemos encontrado, y que hemos tomado como referencia, es la publicada en 1995 por dermatólogos de la Clínica Mayo de Rochester, Estados Unidos, que reunió a 48 pacientes en el período comprendido entre 1980–1992. Más recientemente se han publicado otras series que reúnen un número de pacientes que oscila entre los 2–15 casos25.
Comparando nuestros resultados con los de otras series, destaca la ausencia de predominio femenino, aunque en la serie americana de la clínica Mayo era discreto, con 28 mujeres y 20 varones. Nuestra edad media de aparición de 49 años es similar a las descritas, que se sitúan en torno a los 50 años.
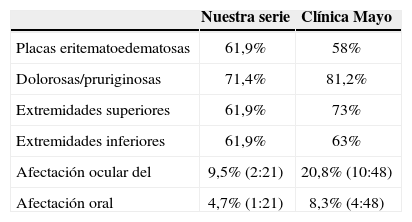
Las lesiones más frecuentemente encontradas se corresponden con las de estudios previos: pápulas y placas eritematoedematosas, que aparecen como únicas lesiones elementales en el 61,9% de nuestros casos y en el 58% de los casos de la serie norteamericana. El 71,4% de los pacientes de nuestra serie y el 81,2% de los pacientes de la serie norteamericana referían las lesiones como dolorosas o pruriginosas. Las localizaciones más frecuentemente encontradas en nuestros casos son casi superponibles a las de los casos de la citada serie: el 61,9 y el 73% en los miembros superiores, el 61,9 y el 63% en los miembros inferiores, respectivamente (tabla 2).
Comparativo de nuestra serie con la serie de la Clínica Mayo
| Nuestra serie | Clínica Mayo | |
| Placas eritematoedematosas | 61,9% | 58% |
| Dolorosas/pruriginosas | 71,4% | 81,2% |
| Extremidades superiores | 61,9% | 73% |
| Extremidades inferiores | 61,9% | 63% |
| Afectación ocular del | 9,5% (2:21) | 20,8% (10:48) |
| Afectación oral | 4,7% (1:21) | 8,3% (4:48) |
Hemos hallado afectación conjuntival en 2 de los 21 casos (epiescleritis) y oral en uno de los 21 casos (ulceración aftosa concomitante). En la serie de la Clínica Mayo aparece también como más frecuente la afectación ocular en 10 de 48 (conjuntivitis en 6, epiescleritis en 2 y ambas en otros 2), seguida de la afectación oral (aftas orales en 4 de 48 pacientes) (tabla 2).
No encontramos afectación de otros órganos en ningún paciente. Uno de nuestros pacientes presentó una neumonía previa a la aparición de las lesiones cutáneas, que respondió a tratamiento antibiótico, por lo que no la consideramos como afectación pulmonar por este proceso, que sí se presentó en 4 de sus 48 pacientes.
El 71,4% de nuestros casos (15 de 21) fue idiopático y encontramos un cuadro hemoproliferativo asociado en 2 pacientes: policitemia vera y un síndrome mielodisplásico concomitante, que a los 5 años desarrolló una leucemia aguda mielogénica. En la serie norteamericana describen una proporción mayor de trastornos hematológicos y oncológicos asociados (26 de 48). Ninguno de nuestros casos se asoció a tumores sólidos (referido en 4 de los 48 de su serie) ni fue inducido por fármacos.
La asociación del síndrome de Sweet con la enfermedad inflamatoria intestinal, como ocurría en uno de nuestros casos, está bien descrita en la literatura médica26. En la serie norteamericana refieren 2 casos de síndrome de Sweet asociado a enfermedad inflamatoria intestinal: uno de enfermedad de Crohn y otro de CU. A diferencia de nuestro caso, que coincidió con un brote de CU, el cuadro cutáneo apareció de forma independiente de la actividad de la enfermedad en su caso de CU, pero también paralelo en el caso de la enfermedad de Crohn.
Los hallazgos de laboratorio más frecuentes coinciden con los de la serie norteamericana: leucocitosis (19/48 versus 10/21) y aumento de la velocidad de sedimentación globular (26/48 versus 7/21).
Con respecto a los tratamientos, los más utilizados en todos los casos recogidos en la literatura médica son los corticoides orales en pauta descendente. Destacamos la utilización en nuestros casos del yoduro potásico, que ocupa el segundo lugar en frecuencia, con el que obtuvimos una respuesta equivalente a la de los corticoides y empleamos fundamentalmente en el tratamiento de las recurrencias.
En el 42,8% de nuestros pacientes tratados con corticoides hubo recurrencias de las lesiones cutáneas tras la suspensión del tratamiento, lo que es discretamente mayor a lo referido en la serie norteamericana (37,5%). Una de nuestras pacientes desarrolló un síndrome de Sweet crónico y precisó tratamiento continuado, que en el momento actual se realiza con colchicina, con buen control.
Limitaciones del estudioEn el estudio realizado se han incluido únicamente los pacientes con síndrome de Sweet a los que se les realizó biopsia cutánea y fue compatible con esta entidad. Por esto, dado que en ocasiones el cuadro clínico típico es suficiente para el inicio del tratamiento con muy buena respuesta (tanto que constituye un criterio diagnóstico) en la mayoría de los casos, sumado a que en ocasiones las lesiones de síndrome de Sweet acaban resolviéndose espontáneamente, es muy probable que el número real de casos sea mucho mayor, y los aquí recogidos sean solo una muestra del total.
ConclusionesEl síndrome de Sweet es una entidad infrecuente pero no rara que aparece predominantemente en sujetos de mediana edad, con localización preferente en las extremidades.
La biopsia cutánea es una herramienta diagnóstica muy rentable y demuestra la presencia de infiltrados dérmicos de polimorfonucleares neutrófilos, generalmente sin cambios a nivel epidérmico, con ausencia de hallazgos de vasculitis leucocitoclástica asociada.
Aunque la mayoría de los casos son idiopáticos, debe descartarse cuidadosamente la posibilidad de que se trate de un cuadro paraneoplásico o asociado a una enfermedad sistémica. Ha de tenerse en cuenta que estos procesos pueden presentarse de forma previa, simultánea o posterior al síndrome de Sweet, por lo que es recomendable un seguimiento prolongado. Debe prestarse especial atención a la detección sistemática de procesos hemoproliferativos. Por último, hay que valorar la posibilidad de que se trate de un síndrome de Sweet inducido por fármacos, y el factor estimulante de colonias de granulocitos es el descrito más frecuentemente.
El tratamiento de elección son los corticoides sistémicos, con tan buena respuesta que constituye un criterio diagnóstico. El yoduro potásico puede considerarse como un tratamiento de primera línea, dada su efectividad, similar a la de los corticoides, y con menores efectos adversos, sobre todo en el tratamiento de las recurrencias.
La etiopatogenia del síndrome de Sweet aún no está aclarada, por lo que se requieren nuevos estudios para tratar de elucidar los mecanismos exactos implicados en ella.
A los doctores J.A. Avilés Izquierdo1, R. Suárez Fernández1, C. Recarte García2, J. Millán Núñez-Cortés2 y M. Lecona Echevarría3, de los servicios de Dermatología1, Medicina Interna2 y Anatomía Patológica3, respectivamente, del Hospital General Universitario Gregorio Marañón de Madrid, sin cuya colaboración no habría sido posible la realización de este trabajo.